library(multinma)
options(mc.cores = parallel::detectCores())#> For execution on a local, multicore CPU with excess RAM we recommend calling
#> options(mc.cores = parallel::detectCores())
#>
#> Attaching package: 'multinma'
#> The following objects are masked from 'package:stats':
#>
#> dgamma, pgamma, qgammaThis vignette describes the analysis of data on the mean off-time
reduction in patients given dopamine agonists as adjunct therapy in
Parkinson’s disease, in a network of 7 trials of 4 active drugs plus
placebo (Dias et al.
2011). The data are available in this package as
parkinsons:
head(parkinsons)
#> studyn trtn y se n diff se_diff
#> 1 1 1 -1.22 0.504 54 NA 0.504
#> 2 1 3 -1.53 0.439 95 -0.31 0.668
#> 3 2 1 -0.70 0.282 172 NA 0.282
#> 4 2 2 -2.40 0.258 173 -1.70 0.382
#> 5 3 1 -0.30 0.505 76 NA 0.505
#> 6 3 2 -2.60 0.510 71 -2.30 0.718We consider analysing these data in three separate ways:
- Using arm-based data (means
yand corresponding standard errorsse); - Using contrast-based data (mean differences
diffand corresponding standard errorsse_diff); - A combination of the two, where some studies contribute arm-based data, and other contribute contrast-based data.
Note: In this case, with Normal likelihoods for both arms and contrasts, we will see that the three analyses give identical results. In general, unless the arm-based likelihood is Normal, results from a model using a contrast-based likelihood will not exactly match those from a model using an arm-based likelihood, since the contrast-based Normal likelihood is only an approximation. Similarity of results depends on the suitability of the Normal approximation, which may not always be appropriate - e.g. with a small number of events or small sample size for a binary outcome. The use of an arm-based likelihood (sometimes called an “exact” likelihood) is therefore preferable where possible in general.
Analysis of arm-based data
We begin with an analysis of the arm-based data - means and standard errors.
Setting up the network
We have arm-level continuous data giving the mean off-time reduction
(y) and standard error (se) in each arm. We
use the function set_agd_arm() to set up the network.
arm_net <- set_agd_arm(parkinsons,
study = studyn,
trt = trtn,
y = y,
se = se,
sample_size = n)
arm_net
#> A network with 7 AgD studies (arm-based).
#>
#> ------------------------------------------------------- AgD studies (arm-based) ----
#> Study Treatment arms
#> 1 2: 1 | 3
#> 2 2: 1 | 2
#> 3 3: 4 | 1 | 2
#> 4 2: 4 | 3
#> 5 2: 4 | 3
#> 6 2: 4 | 5
#> 7 2: 4 | 5
#>
#> Outcome type: continuous
#> ------------------------------------------------------------------------------------
#> Total number of treatments: 5
#> Total number of studies: 7
#> Reference treatment is: 4
#> Network is connectedWe let treatment 4 be set by default as the network reference
treatment, since this results in considerably improved sampling
efficiency over choosing treatment 1 as the network reference. The
sample_size argument is optional, but enables the nodes to
be weighted by sample size in the network plot.
Plot the network structure.
plot(arm_net, weight_edges = TRUE, weight_nodes = TRUE)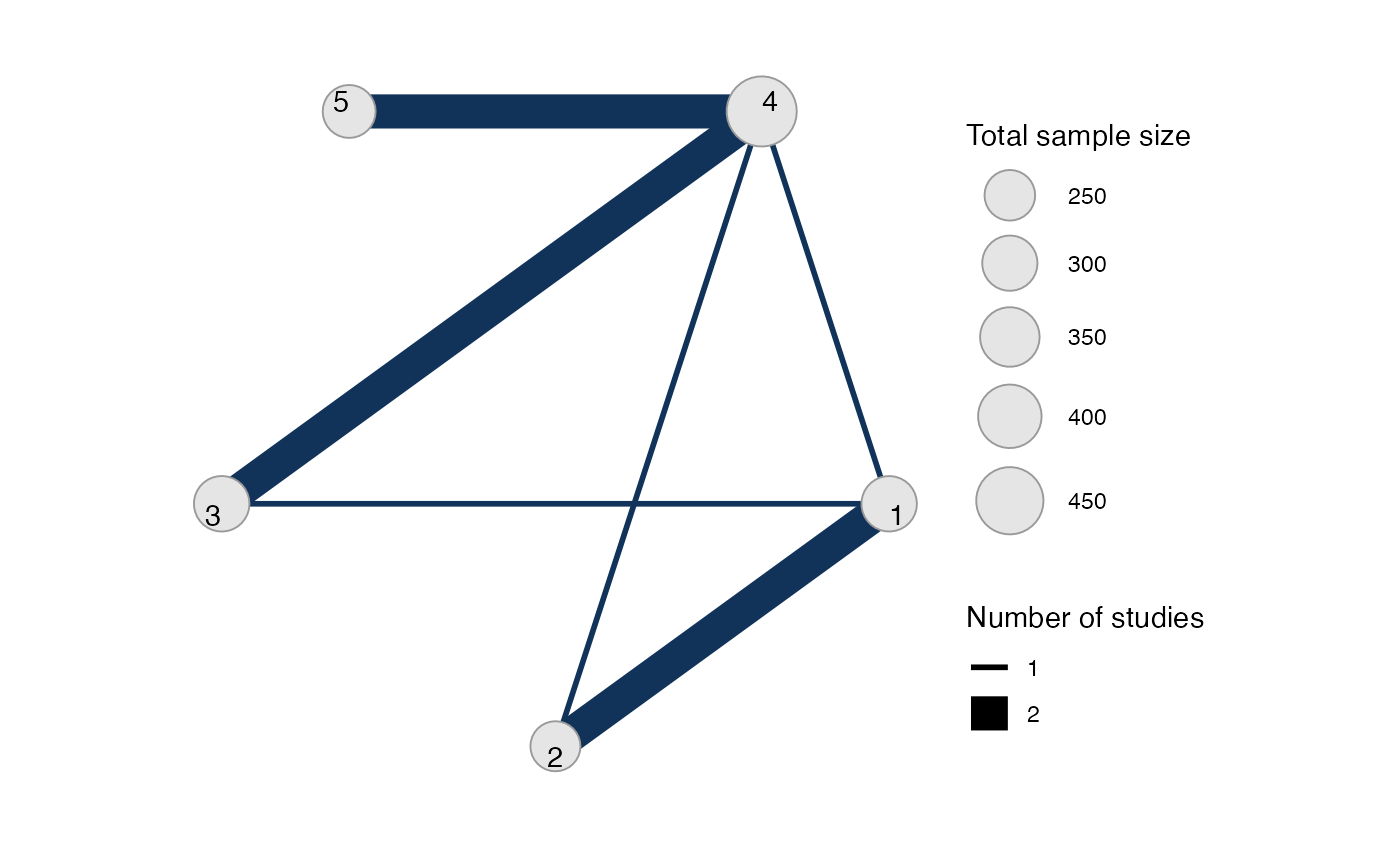
Meta-analysis models
We fit both fixed effect (FE) and random effects (RE) models.
Fixed effect meta-analysis
First, we fit a fixed effect model using the nma()
function with trt_effects = "fixed". We use \mathrm{N}(0, 100^2) prior distributions for
the treatment effects d_k and
study-specific intercepts \mu_j. We can
examine the range of parameter values implied by these prior
distributions with the summary() method:
summary(normal(scale = 100))
#> A Normal prior distribution: location = 0, scale = 100.
#> 50% of the prior density lies between -67.45 and 67.45.
#> 95% of the prior density lies between -196 and 196.The model is fitted using the nma() function.
arm_fit_FE <- nma(arm_net,
trt_effects = "fixed",
prior_intercept = normal(scale = 100),
prior_trt = normal(scale = 10))
#> Note: Setting "4" as the network reference treatment.Basic parameter summaries are given by the print()
method:
arm_fit_FE
#> A fixed effects NMA with a normal likelihood (identity link).
#> Inference for Stan model: normal.
#> 4 chains, each with iter=2000; warmup=1000; thin=1;
#> post-warmup draws per chain=1000, total post-warmup draws=4000.
#>
#> mean se_mean sd 2.5% 25% 50% 75% 97.5% n_eff Rhat
#> d[1] 0.54 0.01 0.47 -0.38 0.23 0.55 0.86 1.47 1503 1
#> d[2] -1.26 0.01 0.52 -2.29 -1.61 -1.26 -0.91 -0.26 1555 1
#> d[3] 0.05 0.01 0.33 -0.60 -0.18 0.04 0.27 0.68 1965 1
#> d[5] -0.30 0.00 0.22 -0.73 -0.45 -0.30 -0.15 0.12 3127 1
#> lp__ -6.76 0.06 2.41 -12.44 -8.12 -6.42 -5.02 -3.12 1724 1
#>
#> Samples were drawn using NUTS(diag_e) at Fri Jun 26 12:37:46 2026.
#> For each parameter, n_eff is a crude measure of effective sample size,
#> and Rhat is the potential scale reduction factor on split chains (at
#> convergence, Rhat=1).By default, summaries of the study-specific intercepts \mu_j are hidden, but could be examined by
changing the pars argument:
The prior and posterior distributions can be compared visually using
the plot_prior_posterior() function:
plot_prior_posterior(arm_fit_FE)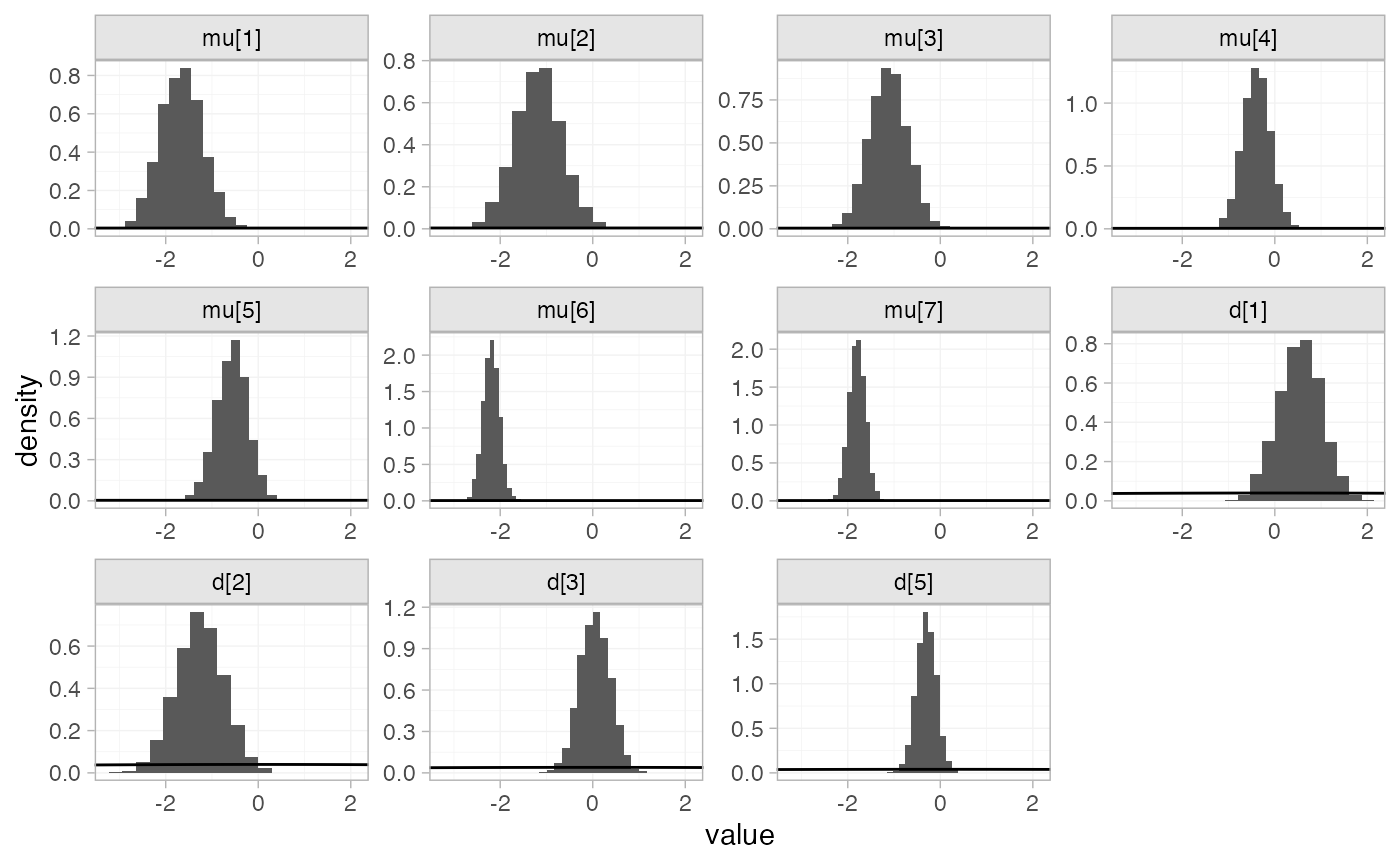
Random effects meta-analysis
We now fit a random effects model using the nma()
function with trt_effects = "random". Again, we use \mathrm{N}(0, 100^2) prior distributions for
the treatment effects d_k and
study-specific intercepts \mu_j, and we
additionally use a \textrm{half-N}(5^2)
prior for the heterogeneity standard deviation \tau. We can examine the range of parameter
values implied by these prior distributions with the
summary() method:
summary(normal(scale = 100))
#> A Normal prior distribution: location = 0, scale = 100.
#> 50% of the prior density lies between -67.45 and 67.45.
#> 95% of the prior density lies between -196 and 196.
summary(half_normal(scale = 5))
#> A half-Normal prior distribution: scale = 5.
#> 50% of the prior density lies between 0 and 3.37.
#> 95% of the prior density lies between 0 and 9.8.Fitting the RE model
arm_fit_RE <- nma(arm_net,
seed = 379394727,
trt_effects = "random",
prior_intercept = normal(scale = 100),
prior_trt = normal(scale = 100),
prior_het = half_normal(scale = 5),
adapt_delta = 0.99)
#> Note: Setting "4" as the network reference treatment.
#> Warning: There were 3 divergent transitions after warmup. See
#> https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
#> to find out why this is a problem and how to eliminate them.
#> Warning: Examine the pairs() plot to diagnose sampling problemsWe do see a small number of divergent transition errors, which cannot
simply be removed by increasing the value of the
adapt_delta argument (by default set to 0.95
for RE models). To diagnose, we use the pairs() method to
investigate where in the posterior distribution these divergences are
happening (indicated by red crosses):
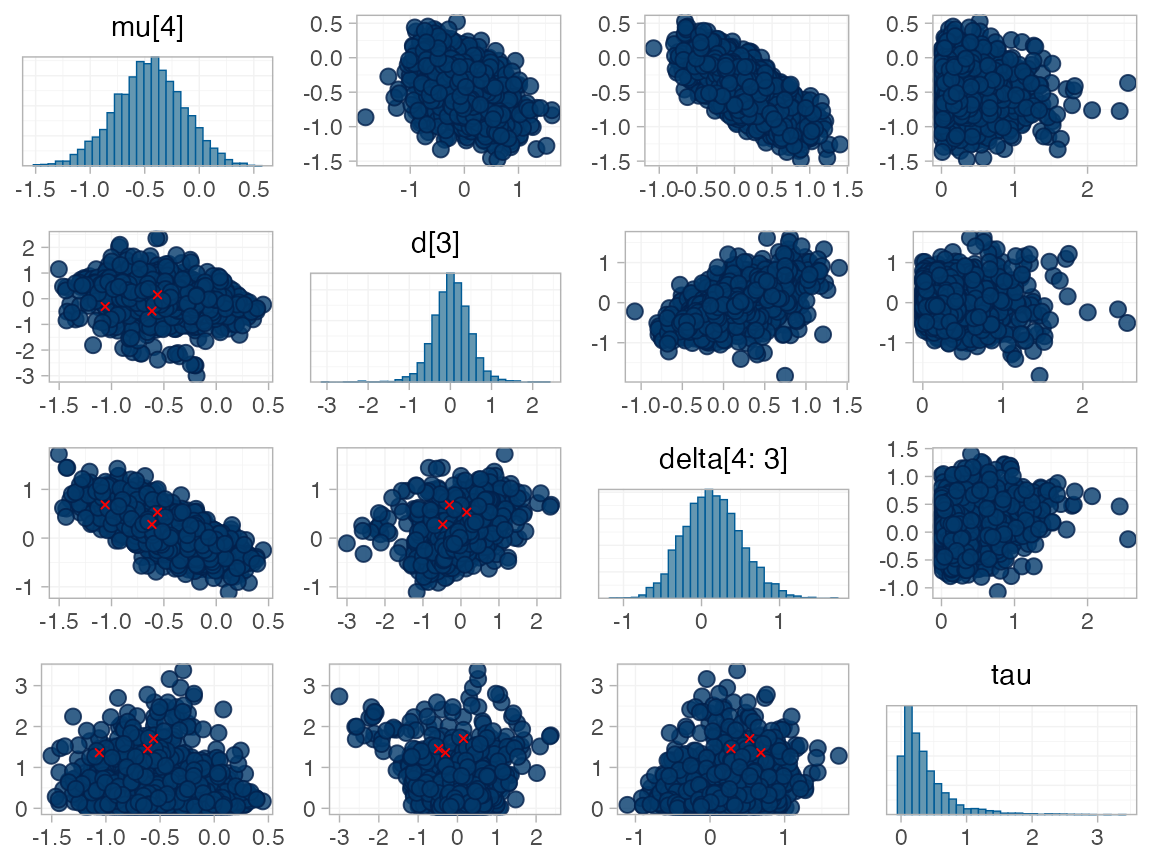
The divergent transitions occur in the upper tail of the heterogeneity standard deviation. In this case, with only a small number of studies, there is not very much information to estimate the heterogeneity standard deviation and the prior distribution may be too heavy-tailed. We could consider a more informative prior distribution for the heterogeneity variance to aid estimation.
Basic parameter summaries are given by the print()
method:
arm_fit_RE
#> A random effects NMA with a normal likelihood (identity link).
#> Inference for Stan model: normal.
#> 4 chains, each with iter=2000; warmup=1000; thin=1;
#> post-warmup draws per chain=1000, total post-warmup draws=4000.
#>
#> mean se_mean sd 2.5% 25% 50% 75% 97.5% n_eff Rhat
#> d[1] 0.53 0.01 0.61 -0.65 0.15 0.55 0.91 1.69 1935 1
#> d[2] -1.33 0.02 0.71 -2.72 -1.75 -1.31 -0.88 -0.05 1448 1
#> d[3] 0.02 0.01 0.47 -0.90 -0.25 0.03 0.31 0.91 1999 1
#> d[5] -0.29 0.01 0.41 -1.09 -0.49 -0.29 -0.10 0.54 2340 1
#> lp__ -13.00 0.10 3.53 -20.90 -15.16 -12.71 -10.46 -7.04 1160 1
#> tau 0.37 0.02 0.39 0.01 0.11 0.26 0.49 1.46 516 1
#>
#> Samples were drawn using NUTS(diag_e) at Fri Jun 26 12:37:49 2026.
#> For each parameter, n_eff is a crude measure of effective sample size,
#> and Rhat is the potential scale reduction factor on split chains (at
#> convergence, Rhat=1).By default, summaries of the study-specific intercepts \mu_j and study-specific relative effects
\delta_{jk} are hidden, but could be
examined by changing the pars argument:
The prior and posterior distributions can be compared visually using
the plot_prior_posterior() function:
plot_prior_posterior(arm_fit_RE)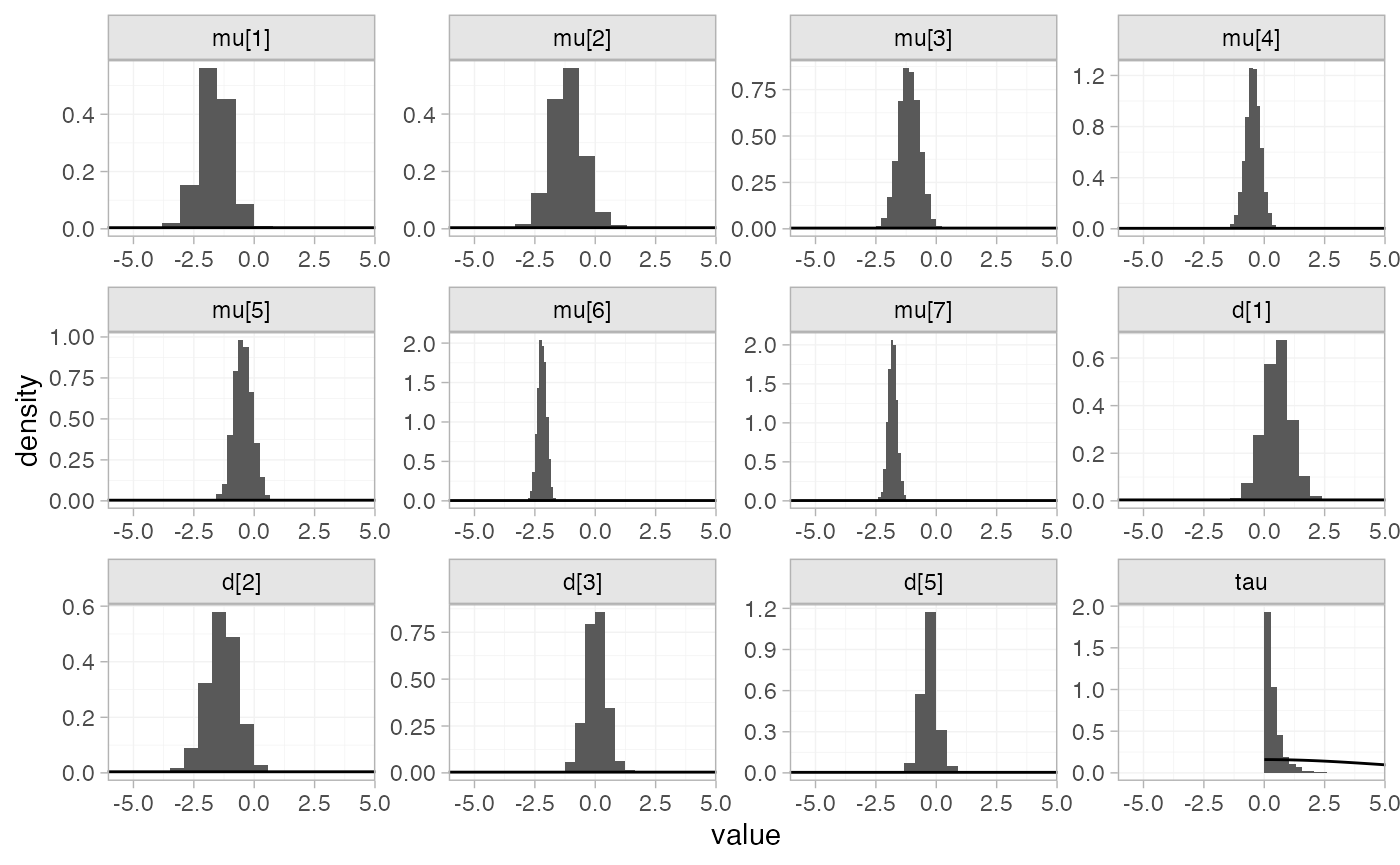
Model comparison
Model fit can be checked using the dic() function:
(arm_dic_FE <- dic(arm_fit_FE))
#> Residual deviance: 13.5 (on 15 data points)
#> pD: 11.2
#> DIC: 24.7
(arm_dic_RE <- dic(arm_fit_RE))
#> Residual deviance: 13.7 (on 15 data points)
#> pD: 12.4
#> DIC: 26.1Both models fit the data well, having posterior mean residual deviance close to the number of data points. The DIC is similar between models, so we choose the FE model based on parsimony.
We can also examine the residual deviance contributions with the
corresponding plot() method.
plot(arm_dic_FE)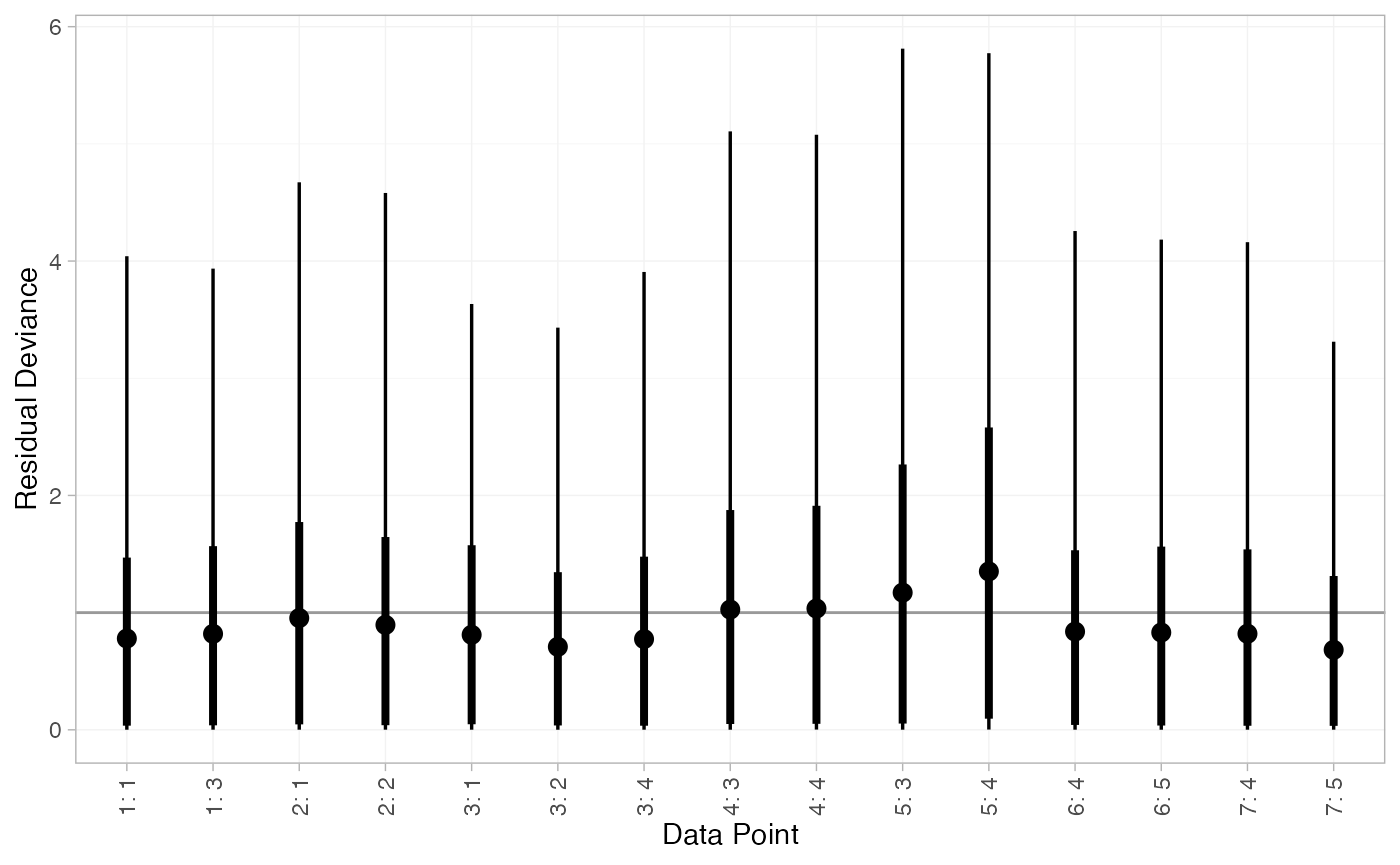
plot(arm_dic_RE)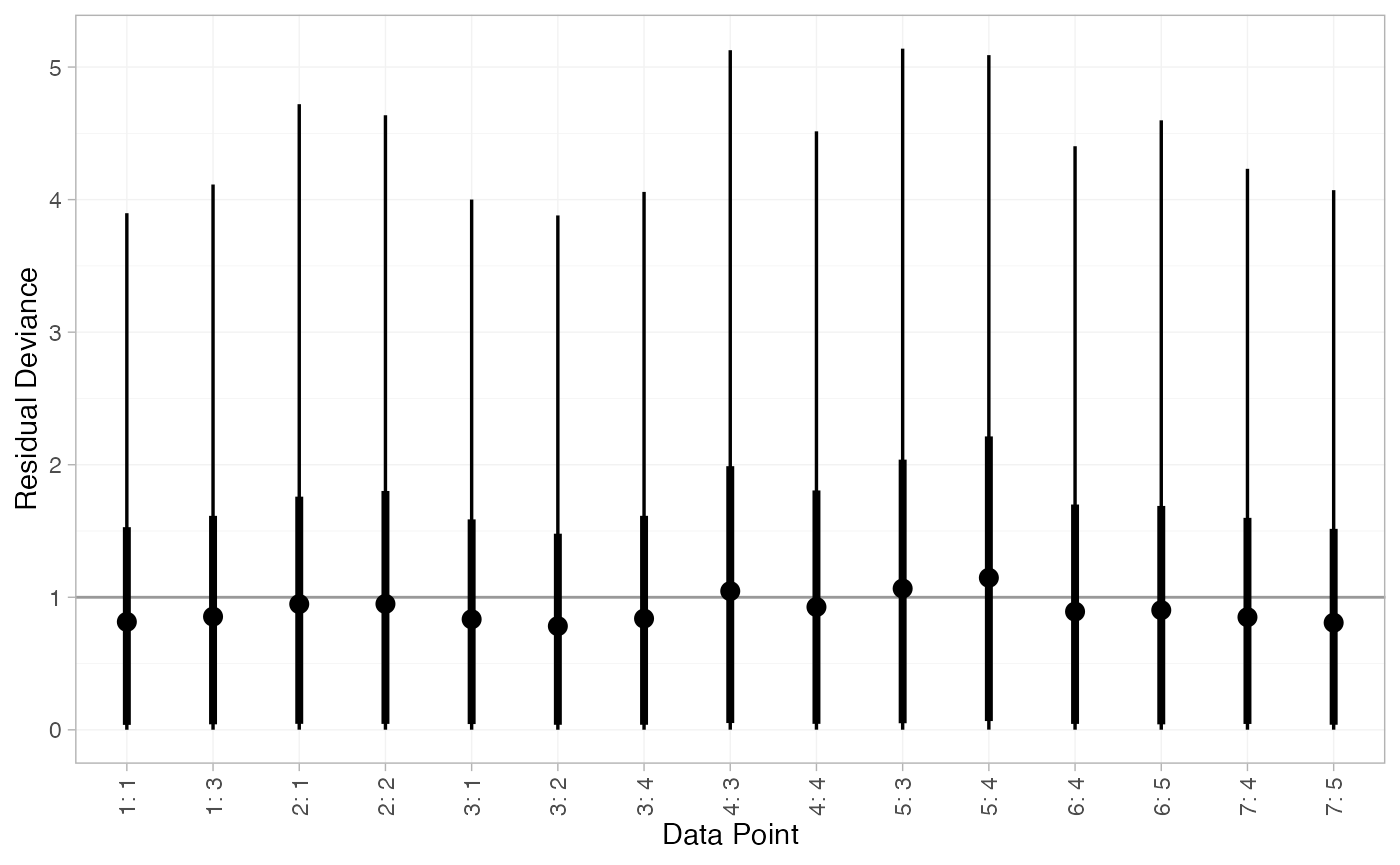
Further results
For comparison with Dias et al. (2011), we can produce relative effects
against placebo using the relative_effects() function with
trt_ref = 1:
(arm_releff_FE <- relative_effects(arm_fit_FE, trt_ref = 1))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> d[4] -0.54 0.47 -1.47 -0.86 -0.55 -0.23 0.38 1520 2166 1
#> d[2] -1.81 0.34 -2.46 -2.05 -1.81 -1.57 -1.13 5565 3253 1
#> d[3] -0.50 0.49 -1.43 -0.82 -0.50 -0.17 0.46 2474 2878 1
#> d[5] -0.84 0.52 -1.85 -1.19 -0.85 -0.50 0.17 1747 2429 1
plot(arm_releff_FE, ref_line = 0)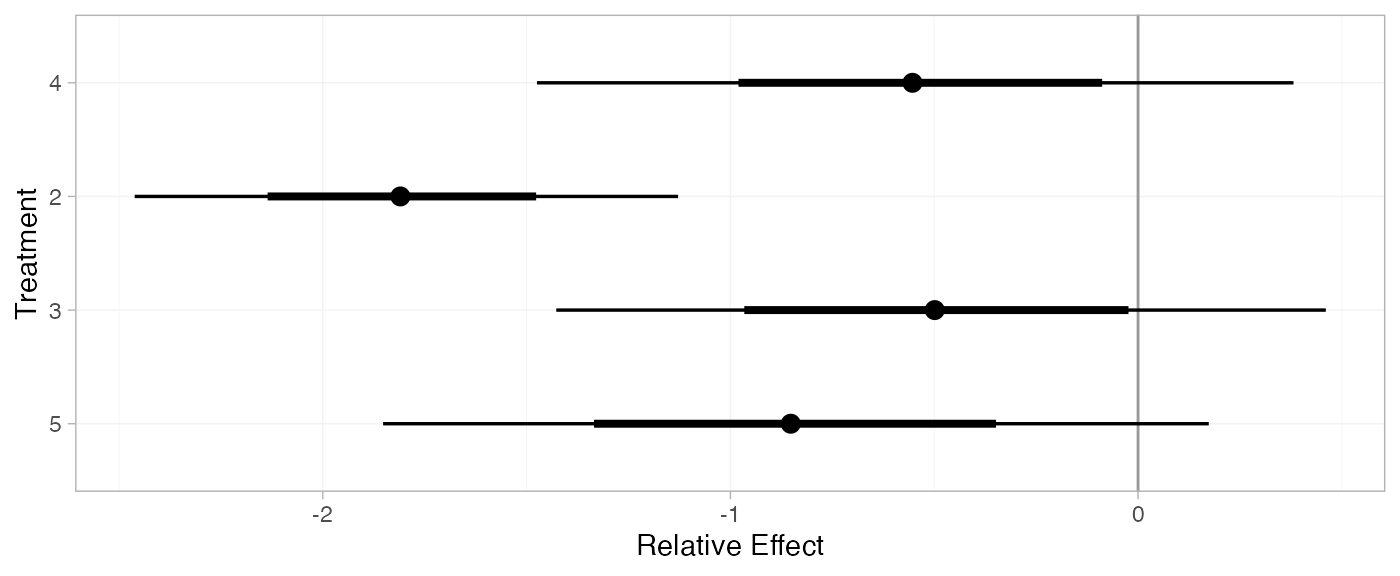
(arm_releff_RE <- relative_effects(arm_fit_RE, trt_ref = 1))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> d[4] -0.53 0.61 -1.69 -0.91 -0.55 -0.15 0.65 1998 1640 1
#> d[2] -1.86 0.54 -2.97 -2.14 -1.84 -1.55 -0.87 4450 1638 1
#> d[3] -0.50 0.64 -1.74 -0.88 -0.50 -0.12 0.72 3033 2509 1
#> d[5] -0.82 0.72 -2.19 -1.25 -0.84 -0.39 0.58 2075 1955 1
plot(arm_releff_RE, ref_line = 0)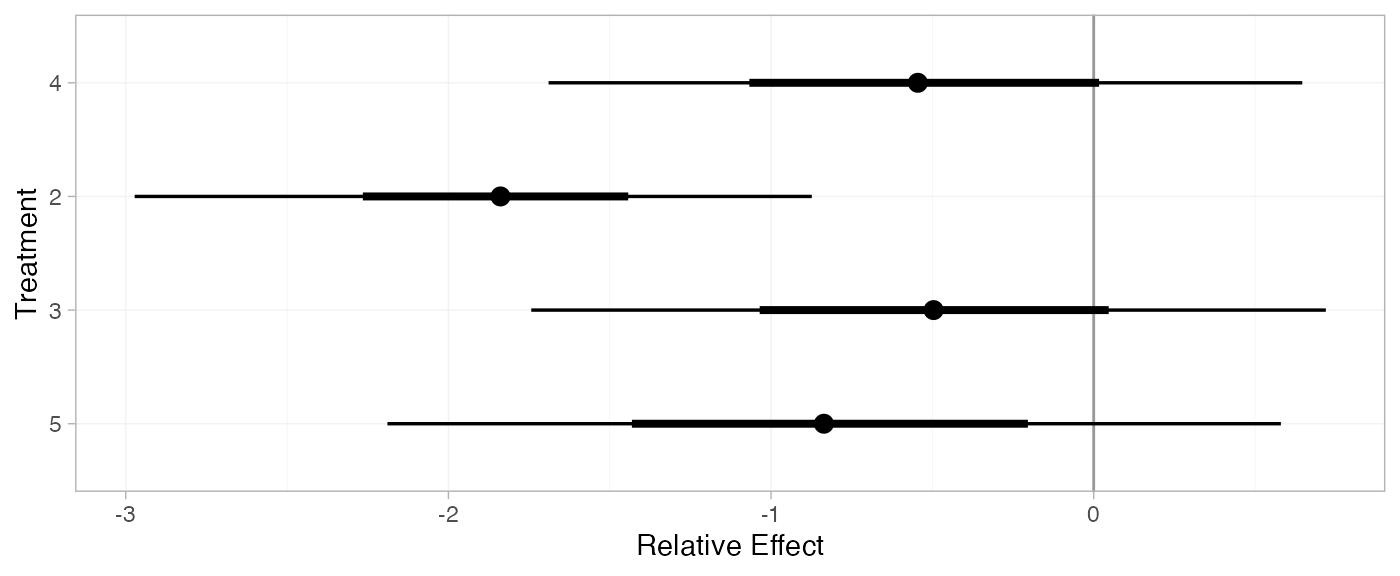
Following Dias et al. (2011), we produce absolute predictions of
the mean off-time reduction on each treatment assuming a Normal
distribution for the outcomes on treatment 1 (placebo) with mean -0.73 and precision 21. We use the predict() method,
where the baseline argument takes a distr()
distribution object with which we specify the corresponding Normal
distribution, and we specify baseline_trt = 1 to indicate
that the baseline distribution corresponds to treatment 1. (Strictly
speaking, type = "response" is unnecessary here, since the
identity link function was used.)
arm_pred_FE <- predict(arm_fit_FE,
baseline = distr(qnorm, mean = -0.73, sd = 21^-0.5),
type = "response",
baseline_trt = 1)
arm_pred_FE
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4] -1.27 0.53 -2.29 -1.62 -1.28 -0.91 -0.24 1701 2585 1
#> pred[1] -0.73 0.22 -1.16 -0.88 -0.73 -0.58 -0.29 3910 3936 1
#> pred[2] -2.53 0.41 -3.33 -2.81 -2.53 -2.26 -1.74 5361 3716 1
#> pred[3] -1.22 0.54 -2.27 -1.59 -1.22 -0.86 -0.17 2650 3202 1
#> pred[5] -1.57 0.57 -2.68 -1.96 -1.58 -1.18 -0.44 1935 2716 1
plot(arm_pred_FE)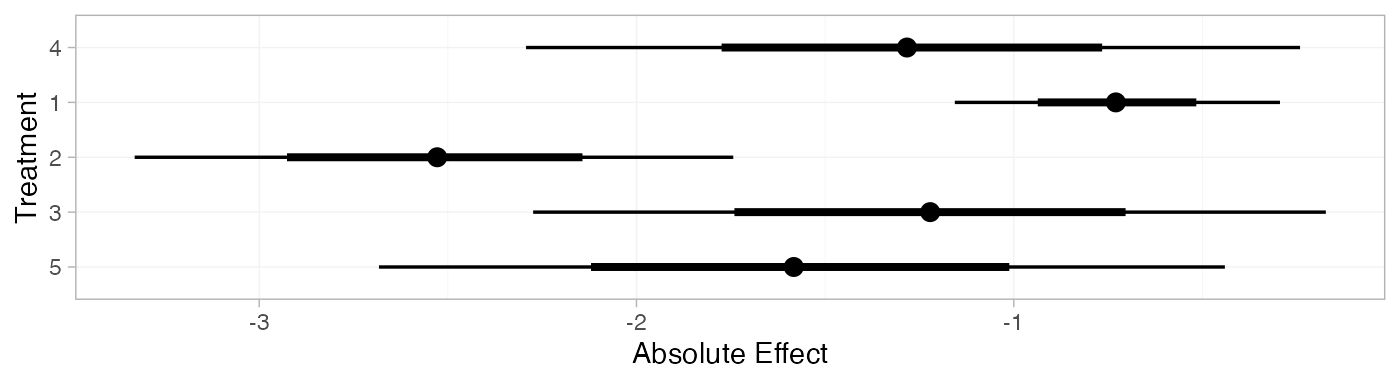
arm_pred_RE <- predict(arm_fit_RE,
baseline = distr(qnorm, mean = -0.73, sd = 21^-0.5),
type = "response",
baseline_trt = 1)
arm_pred_RE
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4] -1.25 0.64 -2.50 -1.68 -1.26 -0.85 -0.02 2148 1954 1
#> pred[1] -0.73 0.22 -1.16 -0.87 -0.73 -0.58 -0.30 3976 3867 1
#> pred[2] -2.58 0.58 -3.76 -2.90 -2.57 -2.25 -1.49 4216 1441 1
#> pred[3] -1.23 0.68 -2.57 -1.63 -1.22 -0.82 0.06 3151 2639 1
#> pred[5] -1.55 0.75 -3.02 -2.00 -1.55 -1.10 -0.08 2181 2142 1
plot(arm_pred_RE)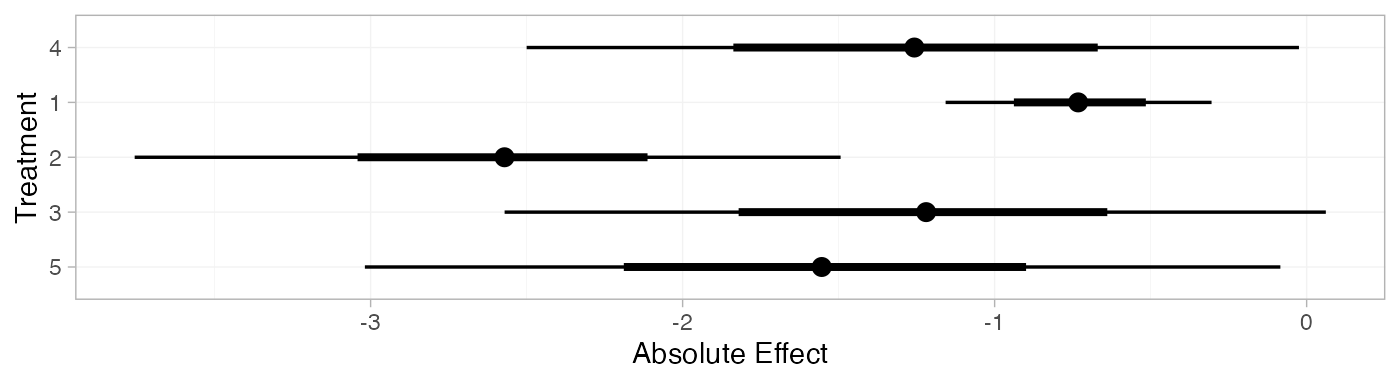
If the baseline argument is omitted, predictions of mean
off-time reduction will be produced for every study in the network based
on their estimated baseline response \mu_j:
arm_pred_FE_studies <- predict(arm_fit_FE, type = "response")
arm_pred_FE_studies
#> ---------------------------------------------------------------------- Study: 1 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[1: 4] -1.66 0.46 -2.52 -1.98 -1.66 -1.35 -0.77 1884 2597 1
#> pred[1: 1] -1.12 0.43 -1.97 -1.40 -1.12 -0.83 -0.27 3777 3318 1
#> pred[1: 2] -2.92 0.52 -3.97 -3.28 -2.92 -2.58 -1.93 3554 3227 1
#> pred[1: 3] -1.61 0.39 -2.35 -1.88 -1.61 -1.35 -0.85 3393 3380 1
#> pred[1: 5] -1.96 0.50 -2.90 -2.32 -1.97 -1.62 -0.95 2052 2630 1
#>
#> ---------------------------------------------------------------------- Study: 2 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[2: 4] -1.18 0.51 -2.16 -1.52 -1.18 -0.85 -0.17 1478 2006 1
#> pred[2: 1] -0.64 0.27 -1.16 -0.82 -0.64 -0.45 -0.12 4690 3456 1
#> pred[2: 2] -2.45 0.24 -2.91 -2.61 -2.45 -2.28 -1.99 5023 3477 1
#> pred[2: 3] -1.14 0.53 -2.17 -1.50 -1.14 -0.78 -0.10 2234 2653 1
#> pred[2: 5] -1.48 0.55 -2.55 -1.86 -1.49 -1.12 -0.38 1704 2387 1
#>
#> ---------------------------------------------------------------------- Study: 3 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[3: 4] -1.12 0.41 -1.93 -1.40 -1.12 -0.85 -0.31 1834 2637 1
#> pred[3: 1] -0.58 0.36 -1.26 -0.82 -0.58 -0.34 0.11 3935 3596 1
#> pred[3: 2] -2.39 0.37 -3.12 -2.64 -2.38 -2.14 -1.66 3721 3110 1
#> pred[3: 3] -1.08 0.48 -2.04 -1.40 -1.08 -0.75 -0.12 2759 2766 1
#> pred[3: 5] -1.43 0.47 -2.34 -1.74 -1.43 -1.11 -0.53 2144 2574 1
#>
#> ---------------------------------------------------------------------- Study: 4 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4: 4] -0.39 0.30 -0.98 -0.60 -0.40 -0.19 0.21 2218 2451 1
#> pred[4: 1] 0.15 0.50 -0.85 -0.19 0.16 0.49 1.11 2290 2536 1
#> pred[4: 2] -1.66 0.56 -2.79 -2.03 -1.64 -1.28 -0.57 2160 2387 1
#> pred[4: 3] -0.35 0.25 -0.83 -0.52 -0.35 -0.18 0.14 5423 3545 1
#> pred[4: 5] -0.70 0.37 -1.42 -0.94 -0.70 -0.45 0.05 2540 2624 1
#>
#> ---------------------------------------------------------------------- Study: 5 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[5: 4] -0.56 0.34 -1.24 -0.79 -0.56 -0.33 0.10 2436 2809 1
#> pred[5: 1] -0.02 0.52 -1.03 -0.38 -0.01 0.33 0.99 2383 2706 1
#> pred[5: 2] -1.83 0.58 -2.94 -2.21 -1.84 -1.44 -0.71 2259 2744 1
#> pred[5: 3] -0.52 0.29 -1.08 -0.72 -0.53 -0.32 0.08 5745 3401 1
#> pred[5: 5] -0.87 0.41 -1.68 -1.14 -0.85 -0.58 -0.08 2827 2930 1
#>
#> ---------------------------------------------------------------------- Study: 6 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[6: 4] -2.20 0.18 -2.56 -2.32 -2.19 -2.08 -1.84 3387 2923 1
#> pred[6: 1] -1.65 0.50 -2.64 -1.99 -1.65 -1.32 -0.65 1650 2421 1
#> pred[6: 2] -3.46 0.55 -4.55 -3.83 -3.46 -3.09 -2.38 1680 2233 1
#> pred[6: 3] -2.15 0.38 -2.90 -2.40 -2.15 -1.89 -1.42 2186 2773 1
#> pred[6: 5] -2.50 0.17 -2.83 -2.61 -2.50 -2.38 -2.16 4987 3117 1
#>
#> ---------------------------------------------------------------------- Study: 7 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[7: 4] -1.80 0.18 -2.16 -1.92 -1.80 -1.68 -1.45 3551 2946 1
#> pred[7: 1] -1.26 0.50 -2.24 -1.59 -1.25 -0.91 -0.29 1679 2355 1
#> pred[7: 2] -3.06 0.55 -4.15 -3.43 -3.06 -2.70 -1.99 1742 2606 1
#> pred[7: 3] -1.75 0.38 -2.50 -2.01 -1.75 -1.50 -1.01 2214 2539 1
#> pred[7: 5] -2.10 0.21 -2.50 -2.24 -2.10 -1.96 -1.70 4914 3210 1
plot(arm_pred_FE_studies)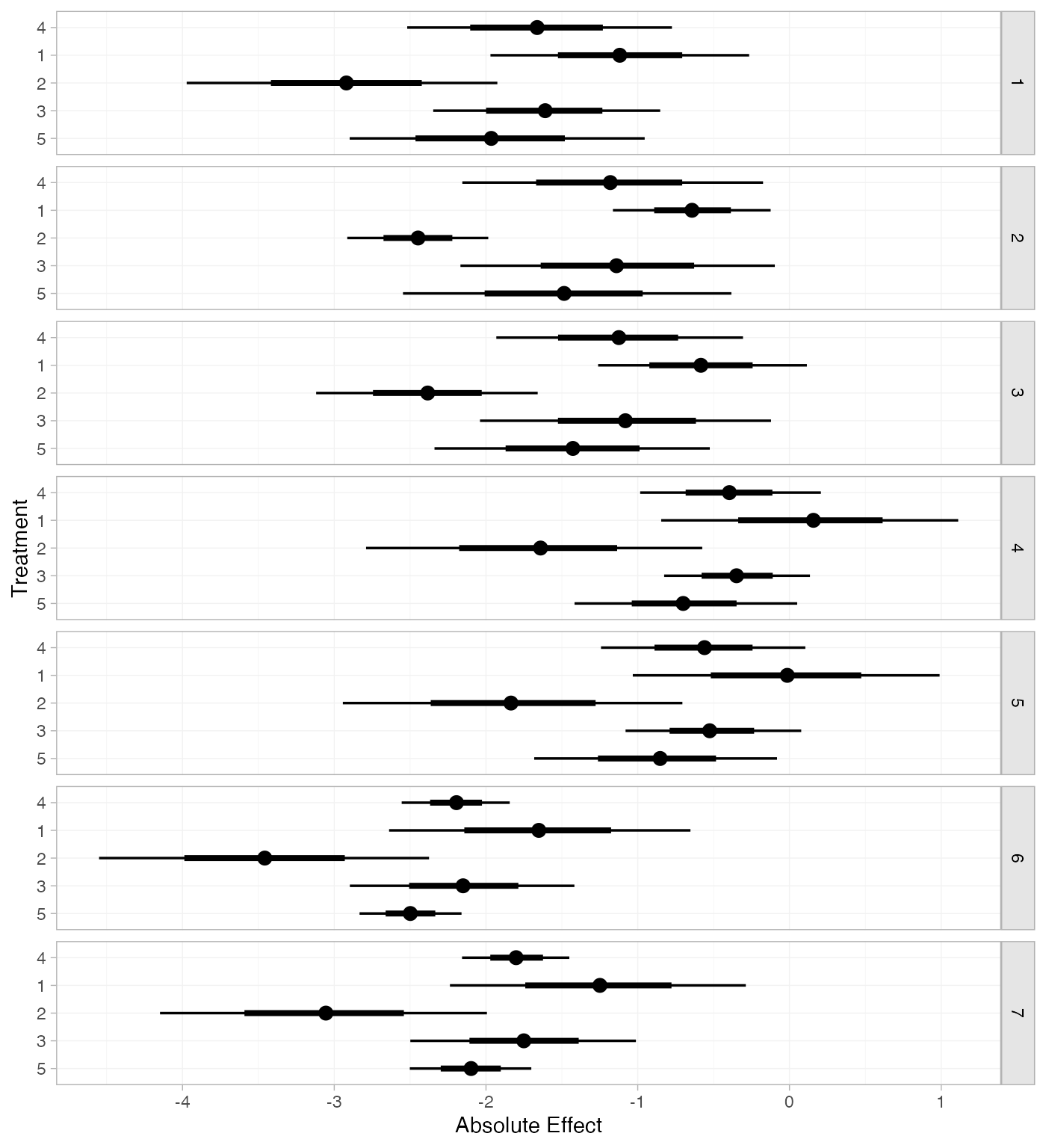
We can also produce treatment rankings, rank probabilities, and cumulative rank probabilities.
(arm_ranks <- posterior_ranks(arm_fit_FE))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> rank[4] 3.49 0.70 2 3 3 4 5 1990 NA 1
#> rank[1] 4.66 0.75 2 5 5 5 5 2335 NA 1
#> rank[2] 1.06 0.30 1 1 1 1 2 2583 2595 1
#> rank[3] 3.51 0.93 2 3 4 4 5 2854 NA 1
#> rank[5] 2.28 0.69 1 2 2 2 4 2545 2543 1
plot(arm_ranks)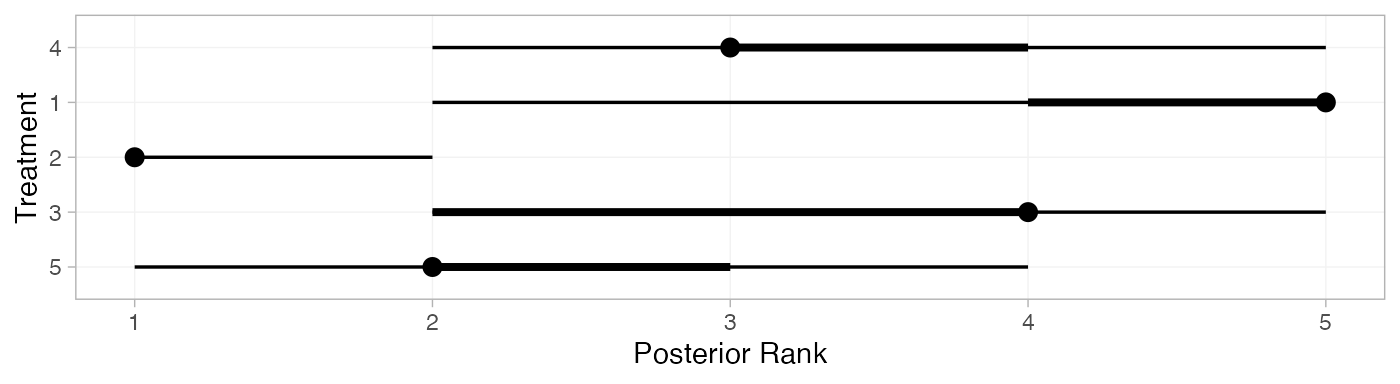
(arm_rankprobs <- posterior_rank_probs(arm_fit_FE))
#> p_rank[1] p_rank[2] p_rank[3] p_rank[4] p_rank[5]
#> d[4] 0.00 0.04 0.49 0.39 0.07
#> d[1] 0.00 0.04 0.06 0.11 0.79
#> d[2] 0.95 0.04 0.01 0.00 0.00
#> d[3] 0.00 0.17 0.26 0.45 0.12
#> d[5] 0.04 0.71 0.18 0.05 0.01
plot(arm_rankprobs)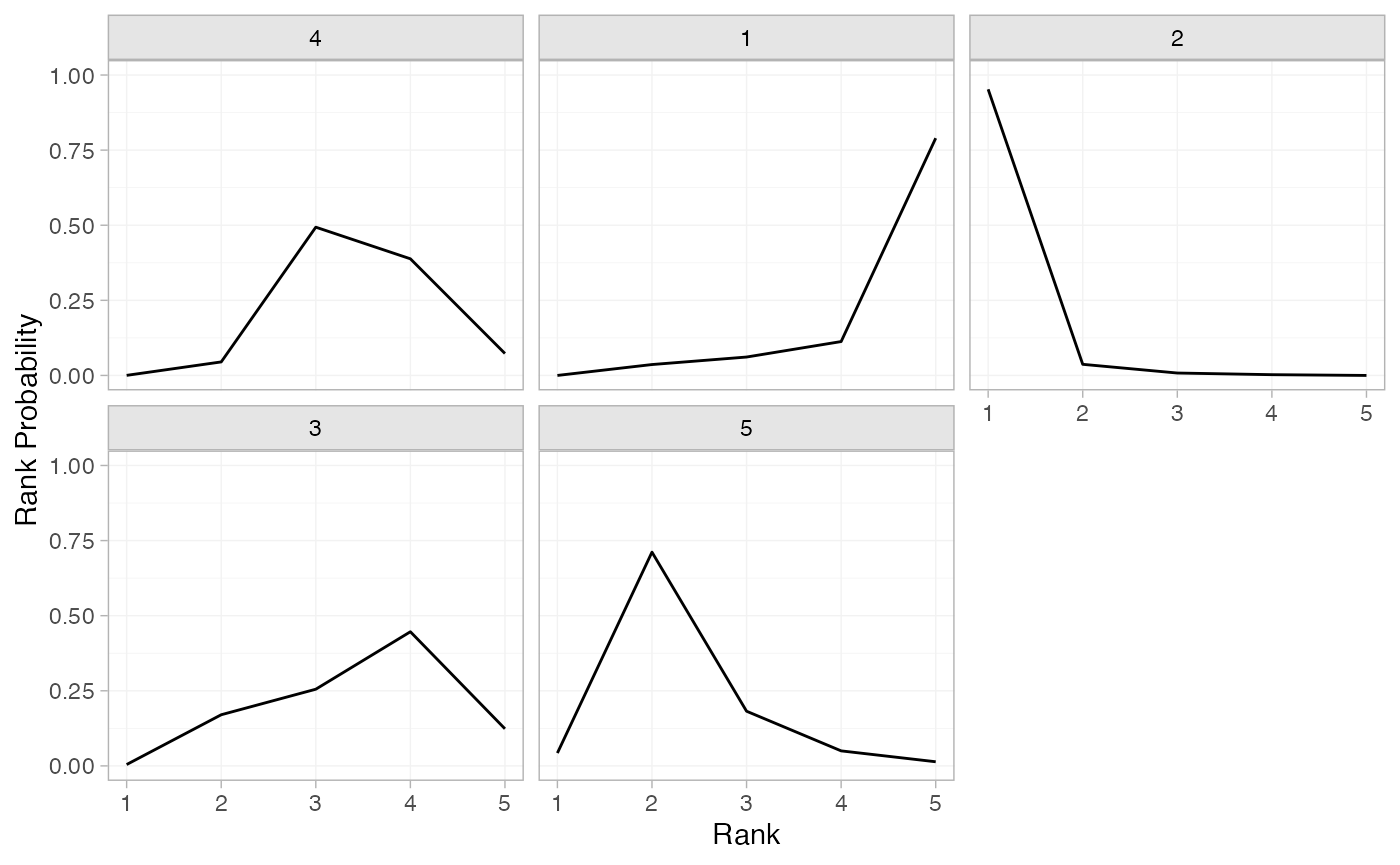
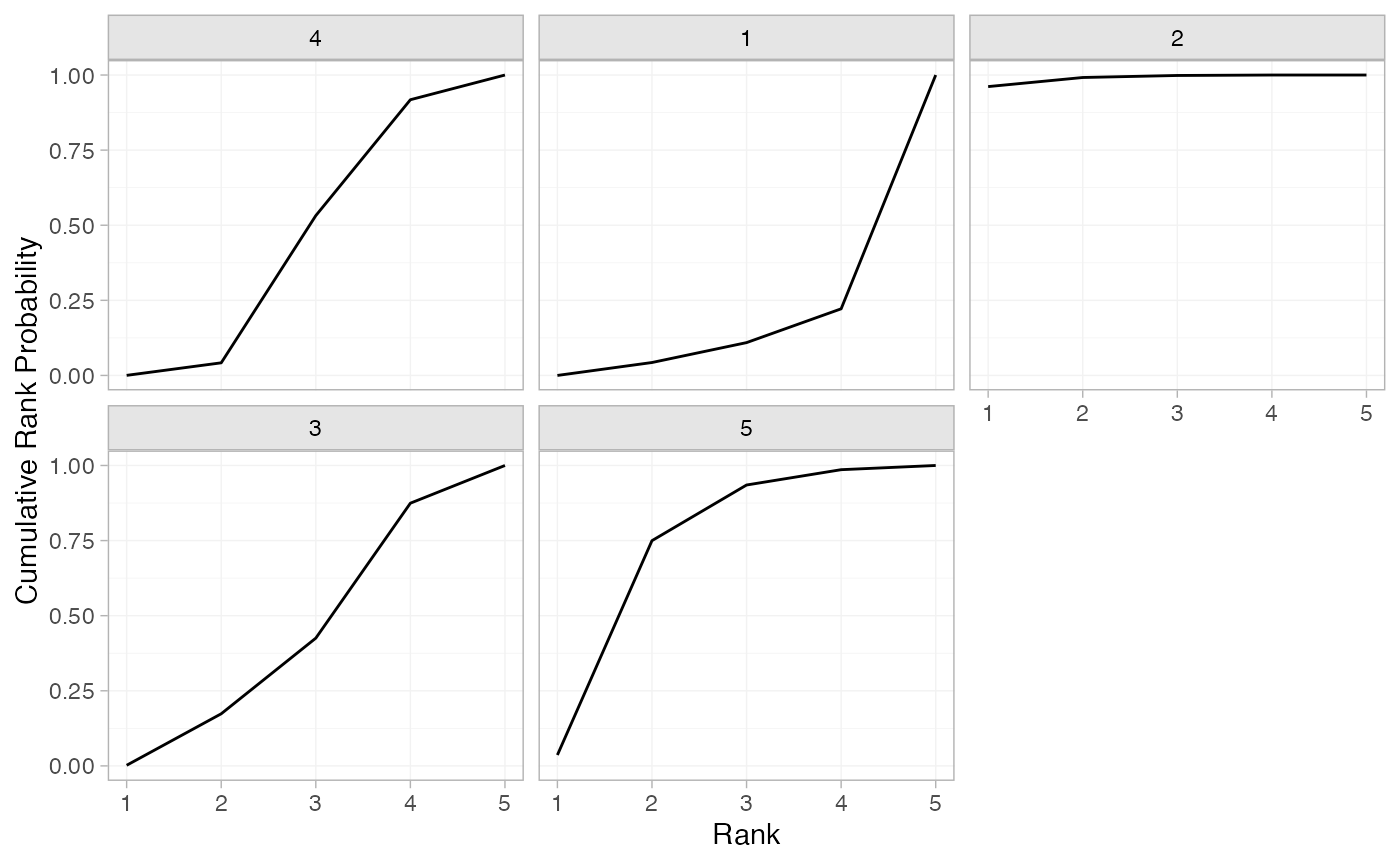
(arm_cumrankprobs <- posterior_rank_probs(arm_fit_FE, cumulative = TRUE))
#> p_rank[1] p_rank[2] p_rank[3] p_rank[4] p_rank[5]
#> d[4] 0.00 0.05 0.54 0.93 1
#> d[1] 0.00 0.04 0.10 0.21 1
#> d[2] 0.95 0.99 1.00 1.00 1
#> d[3] 0.00 0.17 0.43 0.88 1
#> d[5] 0.04 0.75 0.94 0.99 1
plot(arm_cumrankprobs)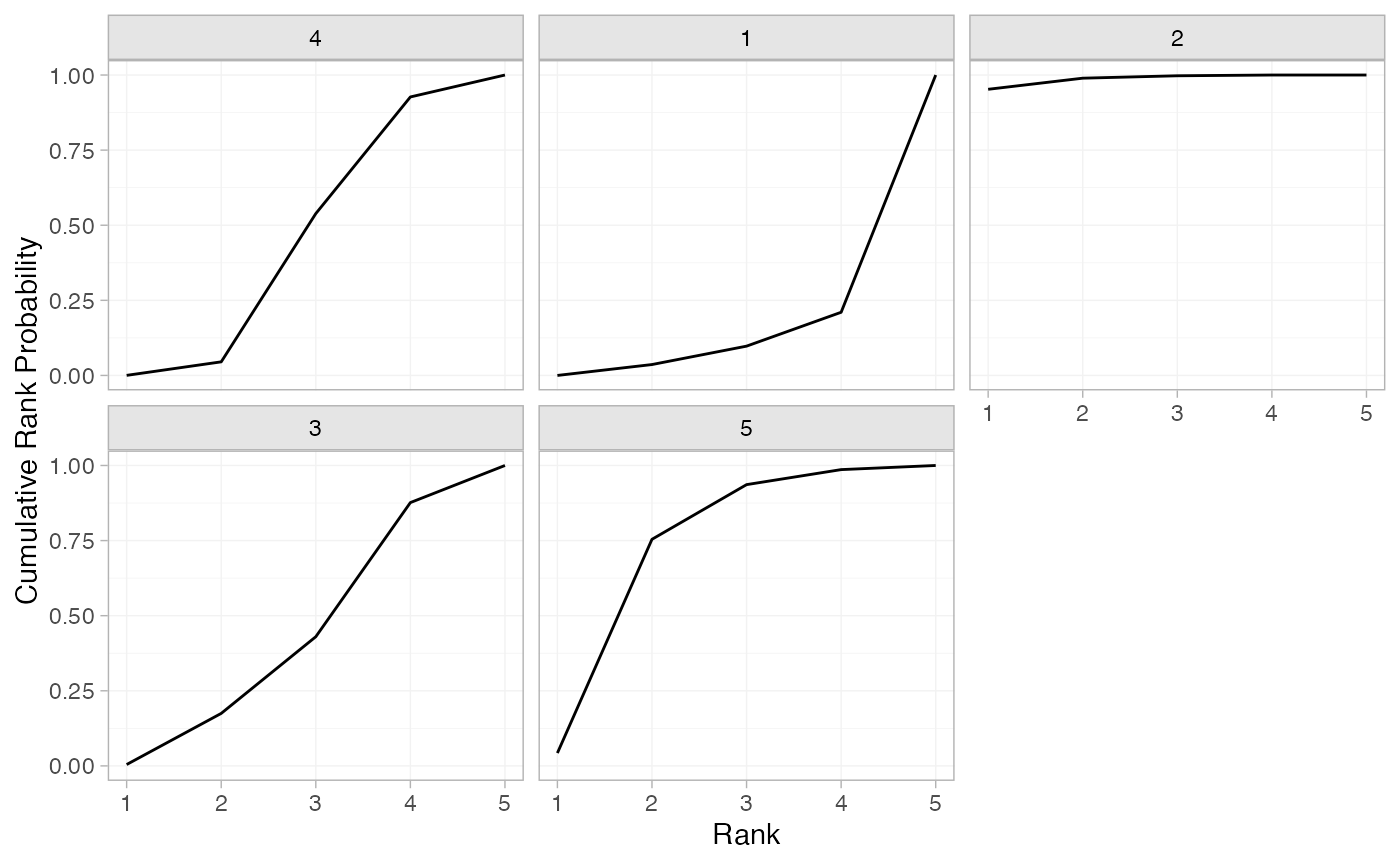
Analysis of contrast-based data
We now perform an analysis using the contrast-based data (mean differences and standard errors).
Setting up the network
With contrast-level data giving the mean difference in off-time
reduction (diff) and standard error (se_diff),
we use the function set_agd_contrast() to set up the
network.
contr_net <- set_agd_contrast(parkinsons,
study = studyn,
trt = trtn,
y = diff,
se = se_diff,
sample_size = n)
contr_net
#> A network with 7 AgD studies (contrast-based).
#>
#> -------------------------------------------------- AgD studies (contrast-based) ----
#> Study Treatment arms
#> 1 2: 1 | 3
#> 2 2: 1 | 2
#> 3 3: 4 | 1 | 2
#> 4 2: 4 | 3
#> 5 2: 4 | 3
#> 6 2: 4 | 5
#> 7 2: 4 | 5
#>
#> Outcome type: continuous
#> ------------------------------------------------------------------------------------
#> Total number of treatments: 5
#> Total number of studies: 7
#> Reference treatment is: 4
#> Network is connectedThe sample_size argument is optional, but enables the
nodes to be weighted by sample size in the network plot.
Plot the network structure.
plot(contr_net, weight_edges = TRUE, weight_nodes = TRUE)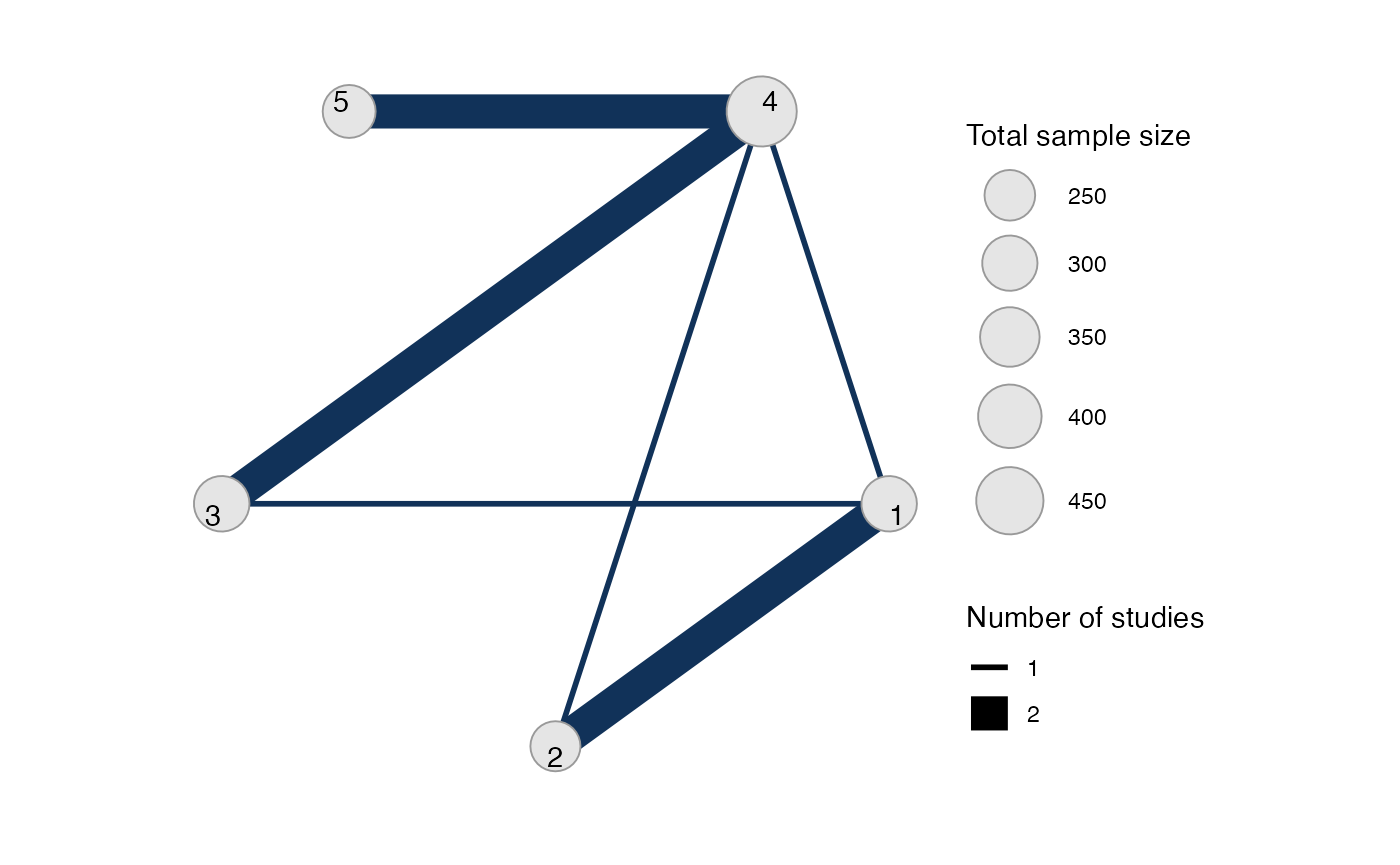
Meta-analysis models
We fit both fixed effect (FE) and random effects (RE) models.
Fixed effect meta-analysis
First, we fit a fixed effect model using the nma()
function with trt_effects = "fixed". We use \mathrm{N}(0, 100^2) prior distributions for
the treatment effects d_k. We can
examine the range of parameter values implied by these prior
distributions with the summary() method:
summary(normal(scale = 100))
#> A Normal prior distribution: location = 0, scale = 100.
#> 50% of the prior density lies between -67.45 and 67.45.
#> 95% of the prior density lies between -196 and 196.The model is fitted using the nma() function.
contr_fit_FE <- nma(contr_net,
trt_effects = "fixed",
prior_trt = normal(scale = 100))
#> Note: Setting "4" as the network reference treatment.Basic parameter summaries are given by the print()
method:
contr_fit_FE
#> A fixed effects NMA with a normal likelihood (identity link).
#> Inference for Stan model: normal.
#> 4 chains, each with iter=2000; warmup=1000; thin=1;
#> post-warmup draws per chain=1000, total post-warmup draws=4000.
#>
#> mean se_mean sd 2.5% 25% 50% 75% 97.5% n_eff Rhat
#> d[1] 0.54 0.01 0.46 -0.37 0.23 0.53 0.84 1.43 2175 1
#> d[2] -1.27 0.01 0.50 -2.26 -1.62 -1.27 -0.93 -0.29 2263 1
#> d[3] 0.05 0.01 0.33 -0.58 -0.18 0.05 0.27 0.71 3058 1
#> d[5] -0.30 0.00 0.21 -0.71 -0.44 -0.30 -0.16 0.08 3831 1
#> lp__ -3.12 0.03 1.36 -6.55 -3.77 -2.82 -2.12 -1.39 1841 1
#>
#> Samples were drawn using NUTS(diag_e) at Fri Jun 26 12:37:59 2026.
#> For each parameter, n_eff is a crude measure of effective sample size,
#> and Rhat is the potential scale reduction factor on split chains (at
#> convergence, Rhat=1).The prior and posterior distributions can be compared visually using
the plot_prior_posterior() function:
plot_prior_posterior(contr_fit_FE)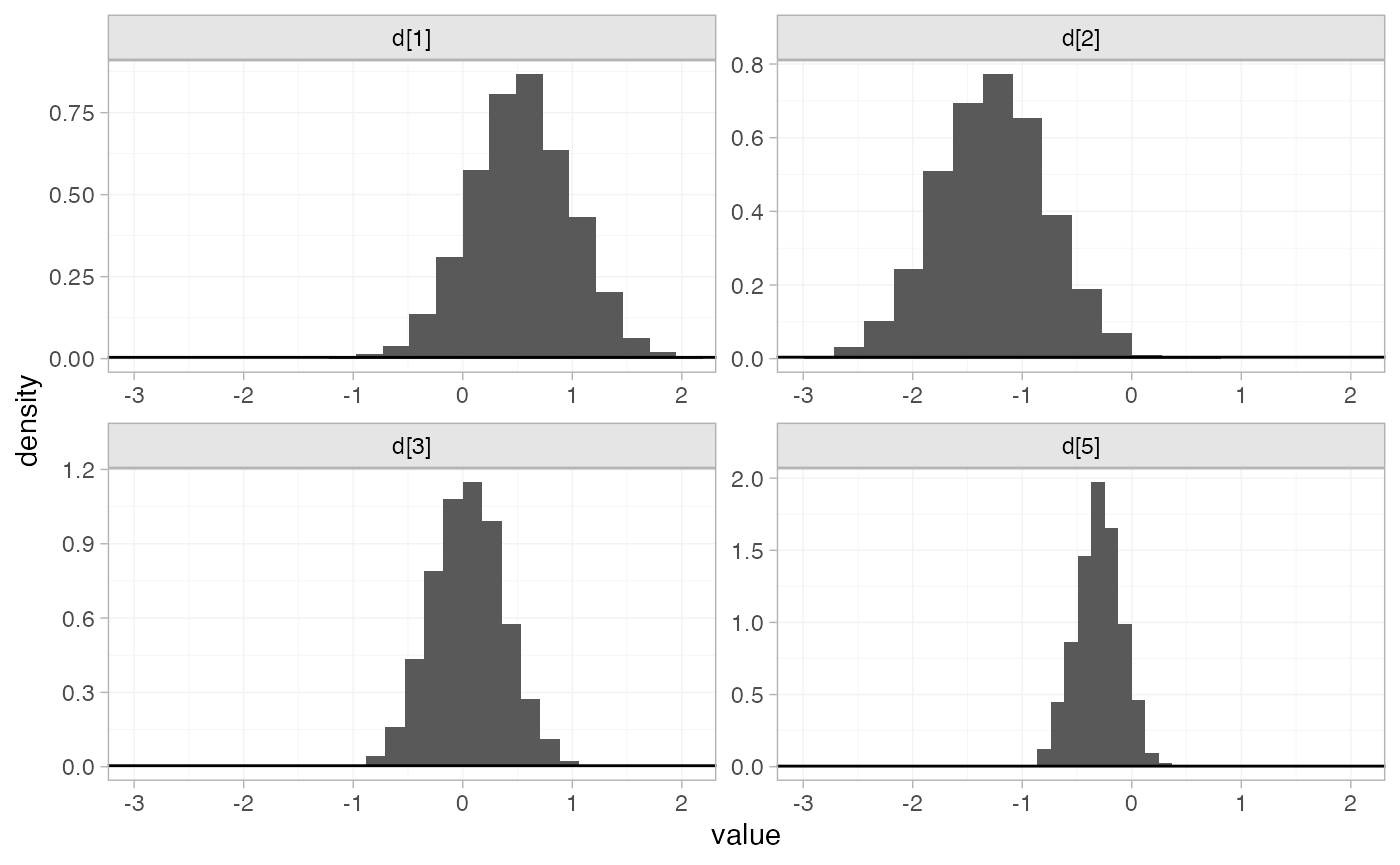
Random effects meta-analysis
We now fit a random effects model using the nma()
function with trt_effects = "random". Again, we use \mathrm{N}(0, 100^2) prior distributions for
the treatment effects d_k, and we
additionally use a \textrm{half-N}(5^2)
prior for the heterogeneity standard deviation \tau. We can examine the range of parameter
values implied by these prior distributions with the
summary() method:
summary(normal(scale = 100))
#> A Normal prior distribution: location = 0, scale = 100.
#> 50% of the prior density lies between -67.45 and 67.45.
#> 95% of the prior density lies between -196 and 196.
summary(half_normal(scale = 5))
#> A half-Normal prior distribution: scale = 5.
#> 50% of the prior density lies between 0 and 3.37.
#> 95% of the prior density lies between 0 and 9.8.Fitting the RE model
contr_fit_RE <- nma(contr_net,
seed = 1150676438,
trt_effects = "random",
prior_trt = normal(scale = 100),
prior_het = half_normal(scale = 5),
adapt_delta = 0.99)
#> Note: Setting "4" as the network reference treatment.We do see a small number of divergent transition errors, which cannot
simply be removed by increasing the value of the
adapt_delta argument (by default set to 0.95
for RE models). To diagnose, we use the pairs() method to
investigate where in the posterior distribution these divergences are
happening (indicated by red crosses):
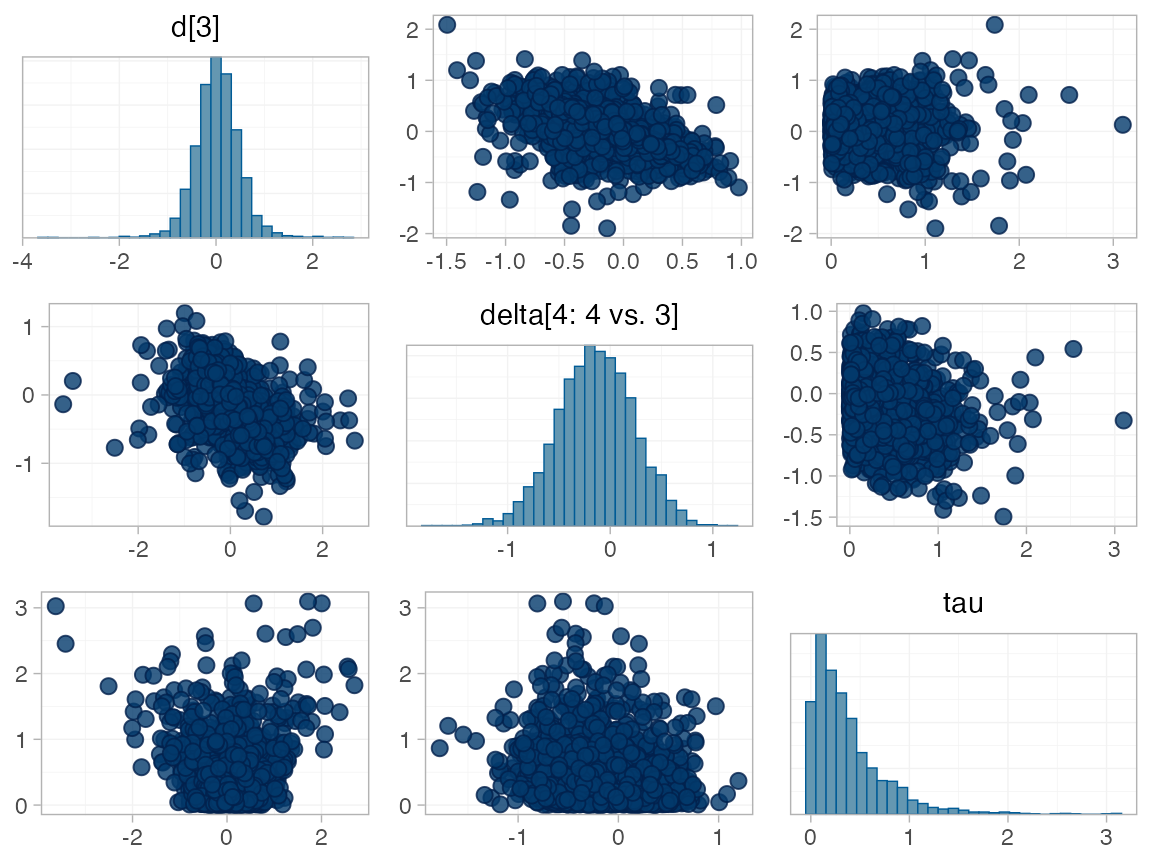
The divergent transitions occur in the upper tail of the heterogeneity standard deviation. In this case, with only a small number of studies, there is not very much information to estimate the heterogeneity standard deviation and the prior distribution may be too heavy-tailed. We could consider a more informative prior distribution for the heterogeneity variance to aid estimation.
Basic parameter summaries are given by the print()
method:
contr_fit_RE
#> A random effects NMA with a normal likelihood (identity link).
#> Inference for Stan model: normal.
#> 4 chains, each with iter=2000; warmup=1000; thin=1;
#> post-warmup draws per chain=1000, total post-warmup draws=4000.
#>
#> mean se_mean sd 2.5% 25% 50% 75% 97.5% n_eff Rhat
#> d[1] 0.51 0.01 0.60 -0.65 0.13 0.52 0.90 1.65 2435 1.00
#> d[2] -1.33 0.01 0.69 -2.70 -1.74 -1.31 -0.89 -0.01 2356 1.00
#> d[3] 0.03 0.01 0.47 -0.83 -0.24 0.03 0.30 0.95 1953 1.00
#> d[5] -0.31 0.01 0.41 -1.19 -0.52 -0.30 -0.10 0.48 2077 1.00
#> lp__ -8.27 0.09 2.92 -14.84 -10.03 -7.97 -6.20 -3.43 1172 1.00
#> tau 0.38 0.01 0.38 0.01 0.12 0.28 0.52 1.42 945 1.01
#>
#> Samples were drawn using NUTS(diag_e) at Fri Jun 26 12:38:01 2026.
#> For each parameter, n_eff is a crude measure of effective sample size,
#> and Rhat is the potential scale reduction factor on split chains (at
#> convergence, Rhat=1).By default, summaries of the study-specific relative effects \delta_{jk} are hidden, but could be examined
by changing the pars argument:
The prior and posterior distributions can be compared visually using
the plot_prior_posterior() function:
plot_prior_posterior(contr_fit_RE)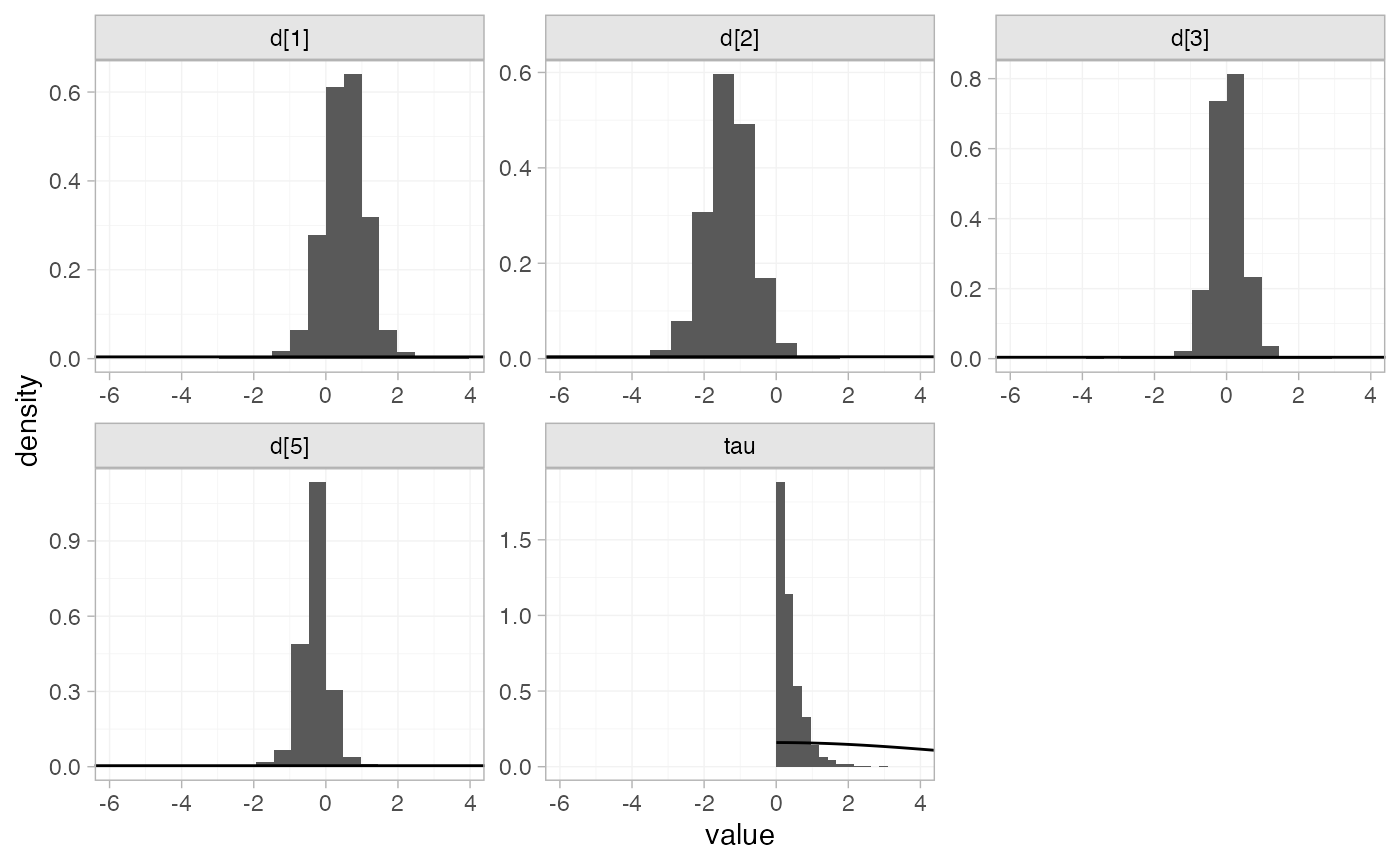
Model comparison
Model fit can be checked using the dic() function:
(contr_dic_FE <- dic(contr_fit_FE))
#> Residual deviance: 6.2 (on 8 data points)
#> pD: 3.9
#> DIC: 10.2
(contr_dic_RE <- dic(contr_fit_RE))
#> Residual deviance: 6.5 (on 8 data points)
#> pD: 5.3
#> DIC: 11.8Both models fit the data well, having posterior mean residual deviance close to the number of data points. The DIC is similar between models, so we choose the FE model based on parsimony.
We can also examine the residual deviance contributions with the
corresponding plot() method.
plot(contr_dic_FE)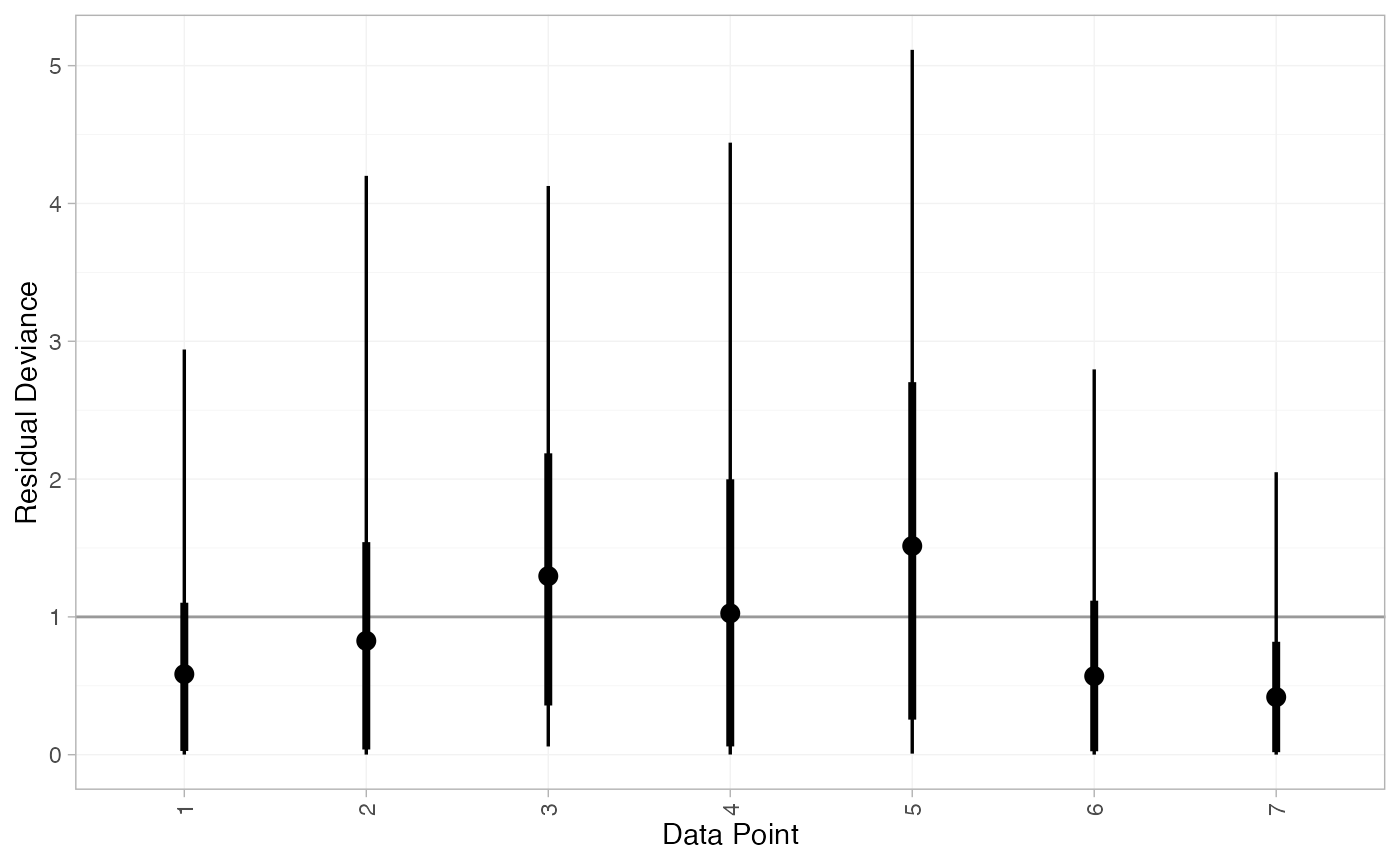
plot(contr_dic_RE)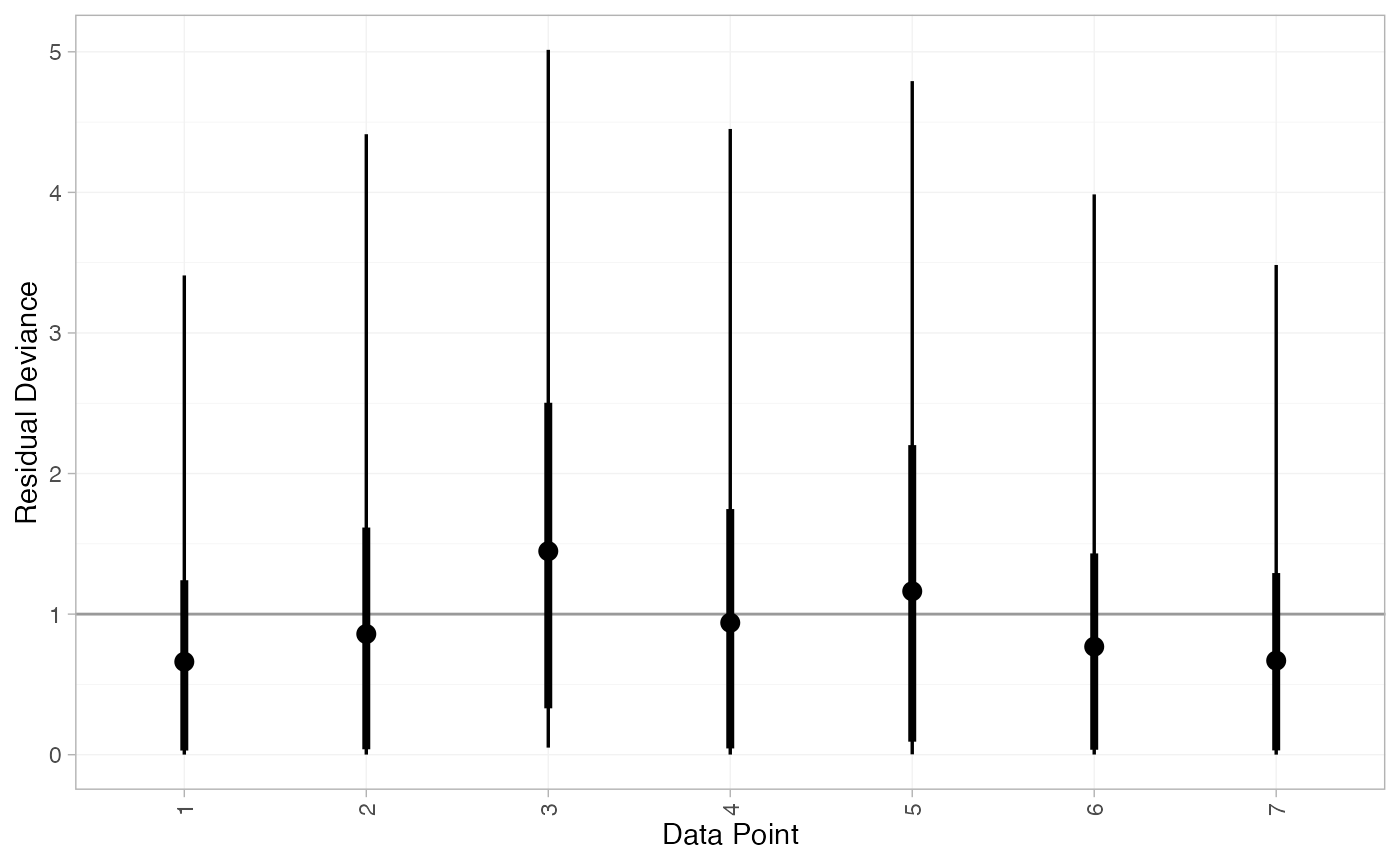
Further results
For comparison with Dias et al. (2011), we can produce relative effects
against placebo using the relative_effects() function with
trt_ref = 1:
(contr_releff_FE <- relative_effects(contr_fit_FE, trt_ref = 1))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> d[4] -0.54 0.46 -1.43 -0.84 -0.53 -0.23 0.37 2193 2485 1
#> d[2] -1.81 0.33 -2.46 -2.03 -1.81 -1.58 -1.17 5584 3277 1
#> d[3] -0.49 0.48 -1.43 -0.81 -0.48 -0.15 0.46 3041 2924 1
#> d[5] -0.83 0.50 -1.83 -1.18 -0.83 -0.50 0.15 2444 2494 1
plot(contr_releff_FE, ref_line = 0)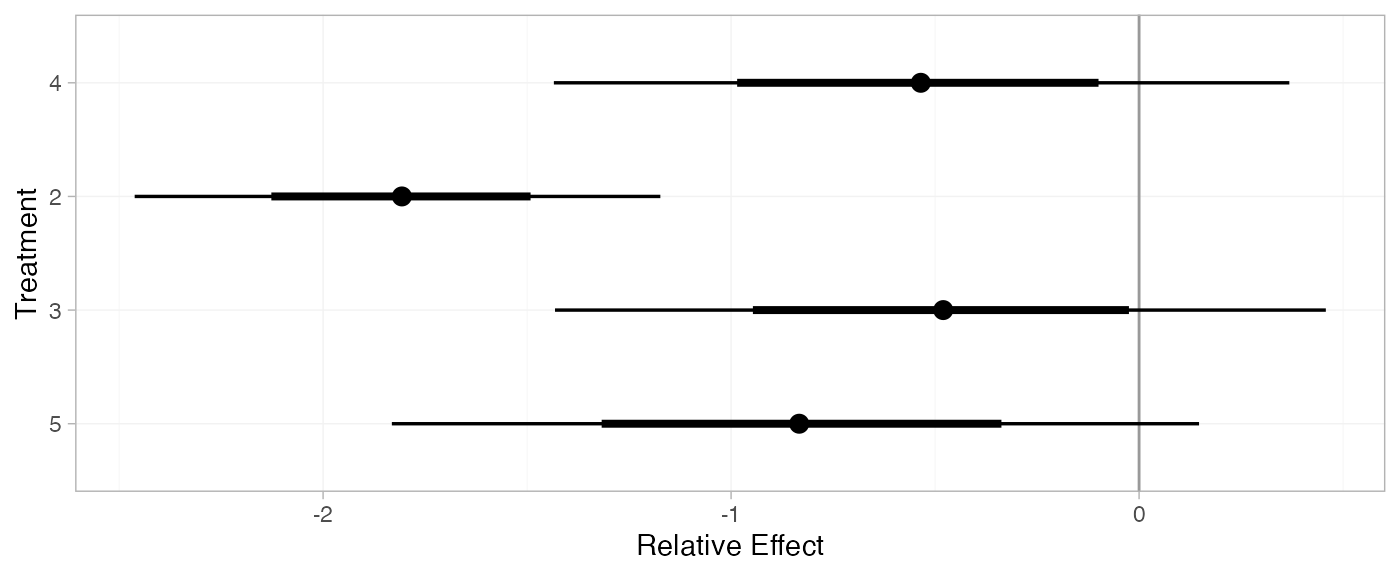
(contr_releff_RE <- relative_effects(contr_fit_RE, trt_ref = 1))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> d[4] -0.51 0.60 -1.65 -0.90 -0.52 -0.13 0.65 2505 2312 1
#> d[2] -1.84 0.51 -2.85 -2.12 -1.83 -1.55 -0.87 3437 2322 1
#> d[3] -0.48 0.63 -1.71 -0.86 -0.49 -0.11 0.75 3210 2479 1
#> d[5] -0.82 0.74 -2.29 -1.25 -0.82 -0.38 0.59 2325 1876 1
plot(contr_releff_RE, ref_line = 0)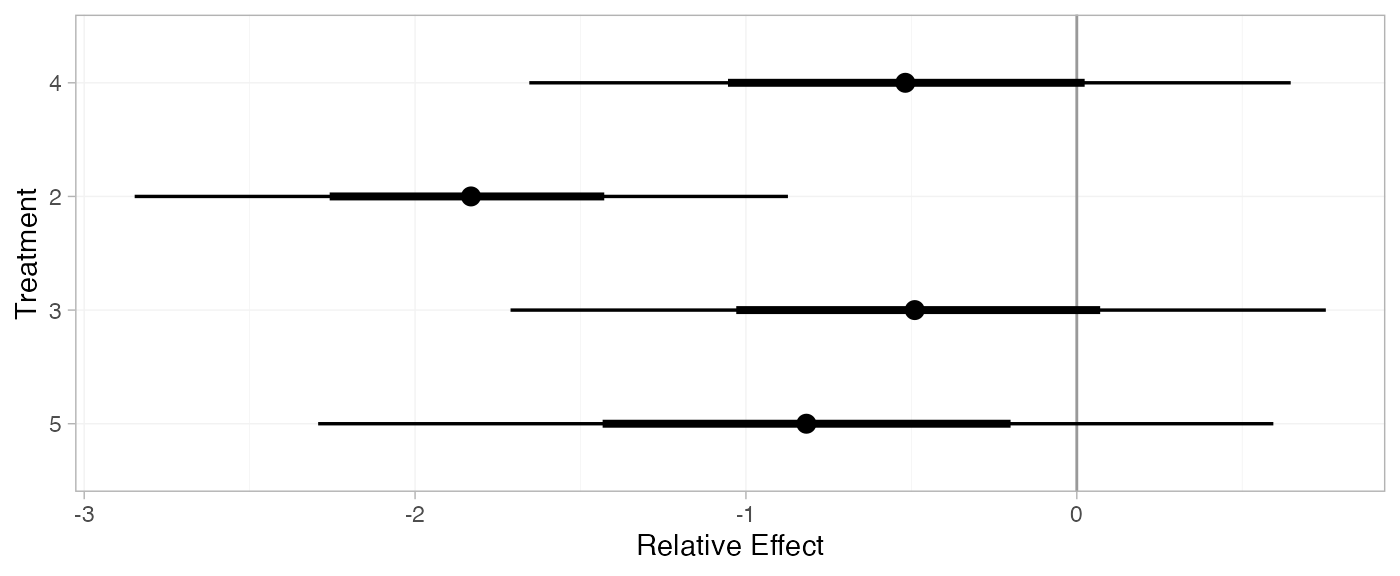
Following Dias et al. (2011), we produce absolute predictions of
the mean off-time reduction on each treatment assuming a Normal
distribution for the outcomes on treatment 1 (placebo) with mean -0.73 and precision 21. We use the predict() method,
where the baseline argument takes a distr()
distribution object with which we specify the corresponding Normal
distribution, and we specify baseline_trt = 1 to indicate
that the baseline distribution corresponds to treatment 1. (Strictly
speaking, type = "response" is unnecessary here, since the
identity link function was used.)
contr_pred_FE <- predict(contr_fit_FE,
baseline = distr(qnorm, mean = -0.73, sd = 21^-0.5),
type = "response",
baseline_trt = 1)
contr_pred_FE
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4] -1.26 0.51 -2.27 -1.61 -1.27 -0.91 -0.25 2343 3040 1
#> pred[1] -0.73 0.22 -1.16 -0.88 -0.73 -0.58 -0.30 3988 3629 1
#> pred[2] -2.54 0.39 -3.33 -2.80 -2.54 -2.28 -1.77 4860 3667 1
#> pred[3] -1.22 0.53 -2.24 -1.58 -1.22 -0.87 -0.18 3029 3222 1
#> pred[5] -1.56 0.55 -2.65 -1.94 -1.56 -1.19 -0.51 2499 2945 1
plot(contr_pred_FE)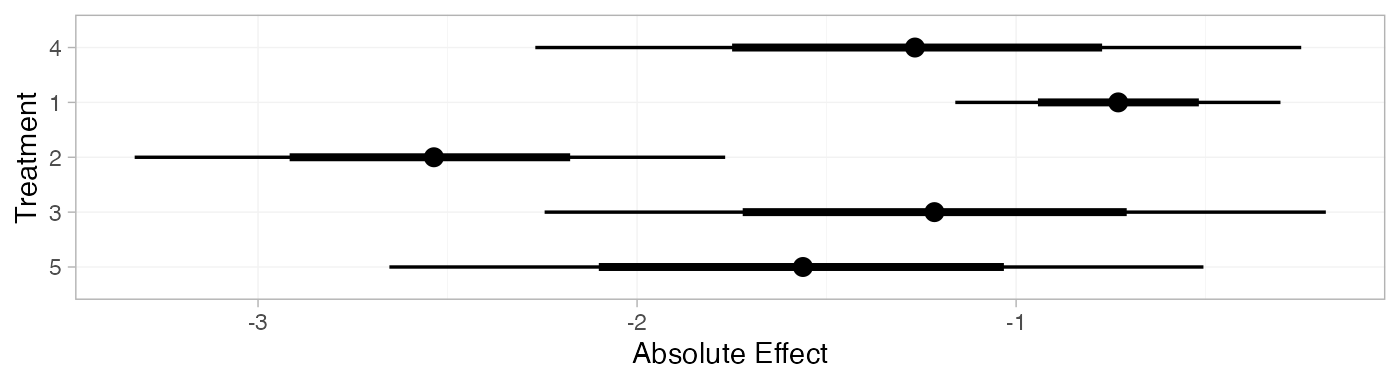
contr_pred_RE <- predict(contr_fit_RE,
baseline = distr(qnorm, mean = -0.73, sd = 21^-0.5),
type = "response",
baseline_trt = 1)
contr_pred_RE
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4] -1.25 0.63 -2.47 -1.65 -1.25 -0.84 -0.01 2628 2267 1
#> pred[1] -0.73 0.22 -1.16 -0.88 -0.73 -0.59 -0.31 3715 3592 1
#> pred[2] -2.58 0.55 -3.67 -2.91 -2.58 -2.24 -1.50 3516 2575 1
#> pred[3] -1.22 0.67 -2.51 -1.63 -1.22 -0.80 0.09 3217 2295 1
#> pred[5] -1.56 0.77 -3.09 -2.02 -1.55 -1.09 -0.04 2445 2093 1
plot(contr_pred_RE)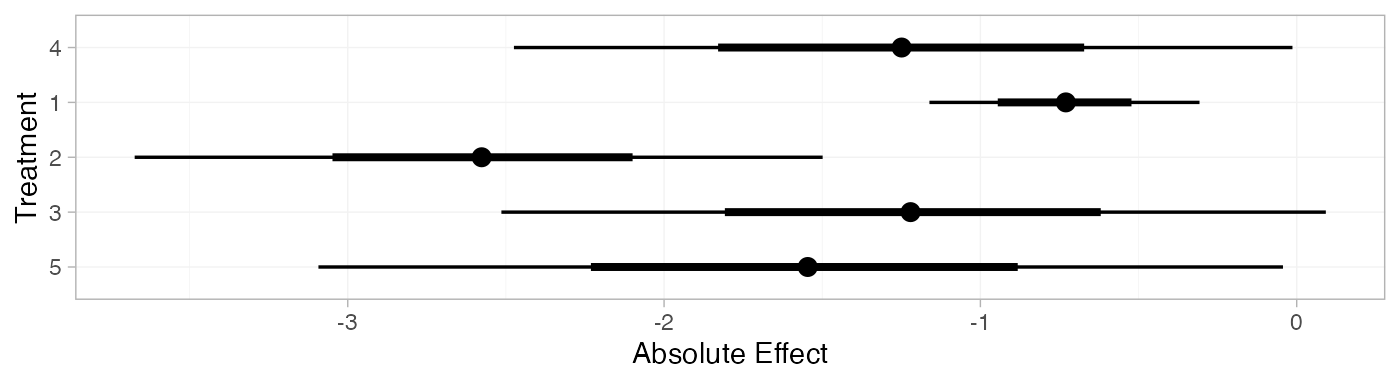
If the baseline argument is omitted an error will be
raised, as there are no study baselines estimated in this network.
# Not run
predict(contr_fit_FE, type = "response")We can also produce treatment rankings, rank probabilities, and cumulative rank probabilities.
(contr_ranks <- posterior_ranks(contr_fit_FE))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> rank[4] 3.49 0.70 2 3 3 4 5 2741 NA 1
#> rank[1] 4.68 0.73 2 5 5 5 5 2532 NA 1
#> rank[2] 1.04 0.23 1 1 1 1 2 2825 2834 1
#> rank[3] 3.51 0.92 2 3 4 4 5 3397 NA 1
#> rank[5] 2.28 0.65 1 2 2 2 4 2845 2127 1
plot(contr_ranks)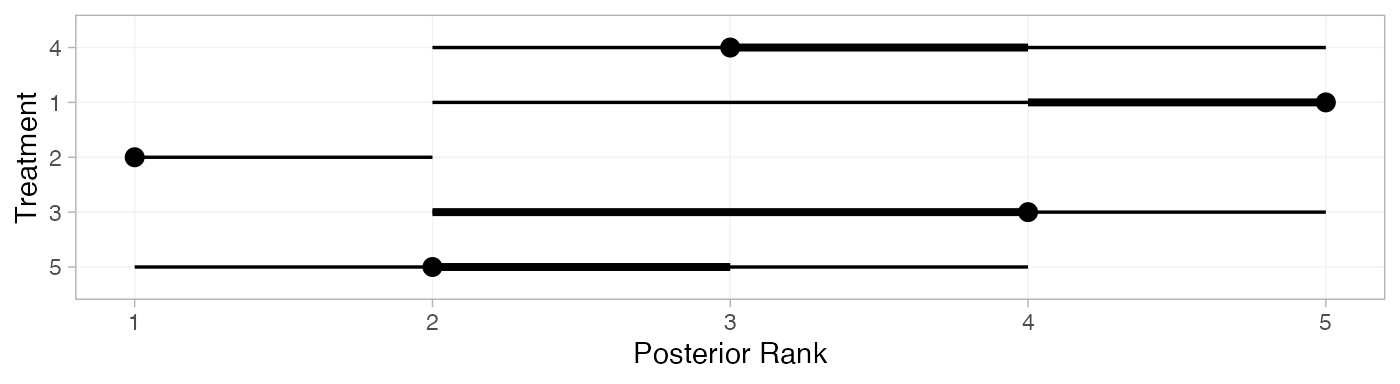
(contr_rankprobs <- posterior_rank_probs(contr_fit_FE))
#> p_rank[1] p_rank[2] p_rank[3] p_rank[4] p_rank[5]
#> d[4] 0.00 0.04 0.50 0.39 0.07
#> d[1] 0.00 0.03 0.06 0.11 0.80
#> d[2] 0.96 0.03 0.00 0.00 0.00
#> d[3] 0.00 0.17 0.25 0.45 0.12
#> d[5] 0.03 0.72 0.19 0.05 0.01
plot(contr_rankprobs)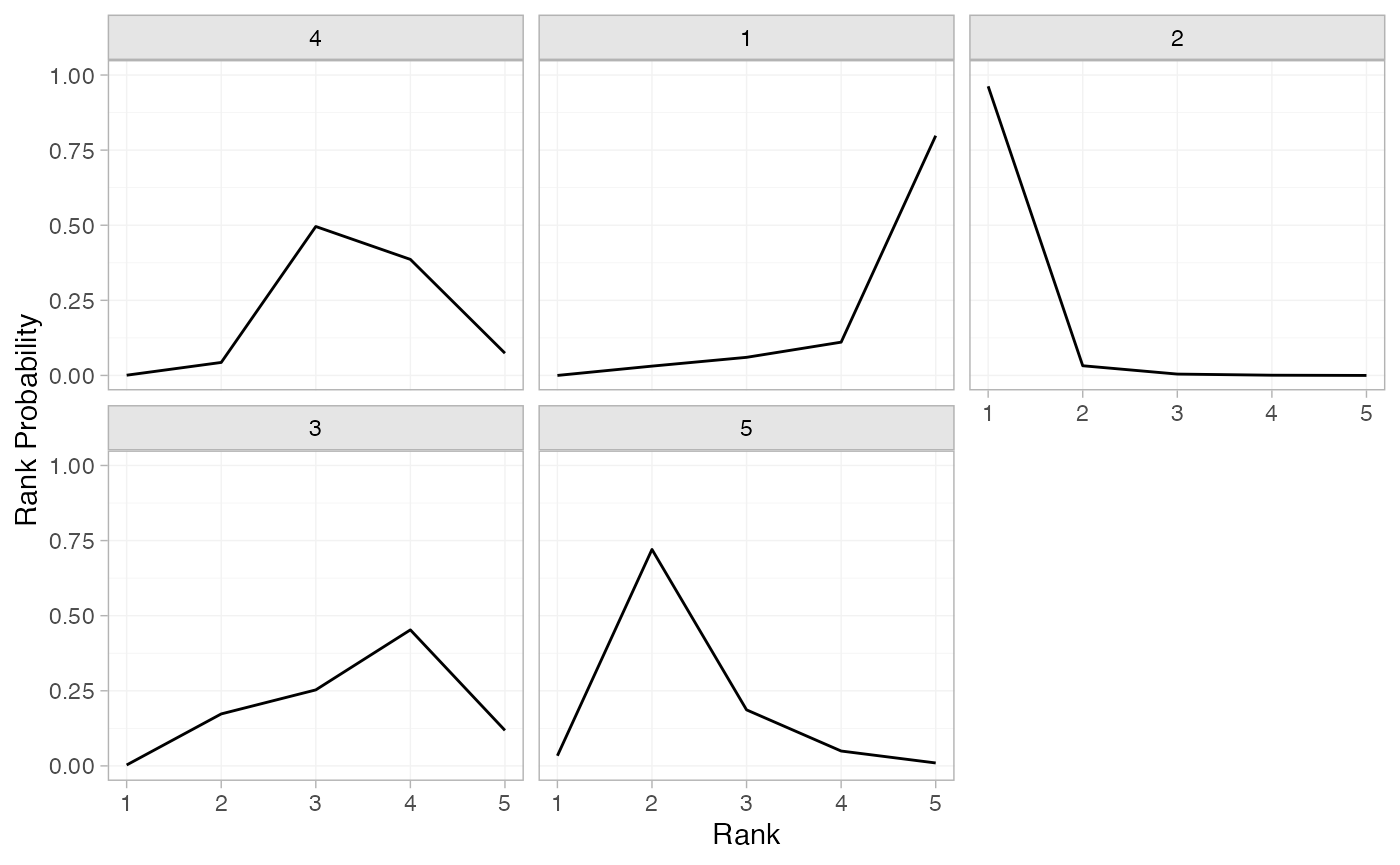
(contr_cumrankprobs <- posterior_rank_probs(contr_fit_FE, cumulative = TRUE))
#> p_rank[1] p_rank[2] p_rank[3] p_rank[4] p_rank[5]
#> d[4] 0.00 0.04 0.54 0.93 1
#> d[1] 0.00 0.03 0.09 0.20 1
#> d[2] 0.96 0.99 1.00 1.00 1
#> d[3] 0.00 0.18 0.43 0.88 1
#> d[5] 0.03 0.75 0.94 0.99 1
plot(contr_cumrankprobs)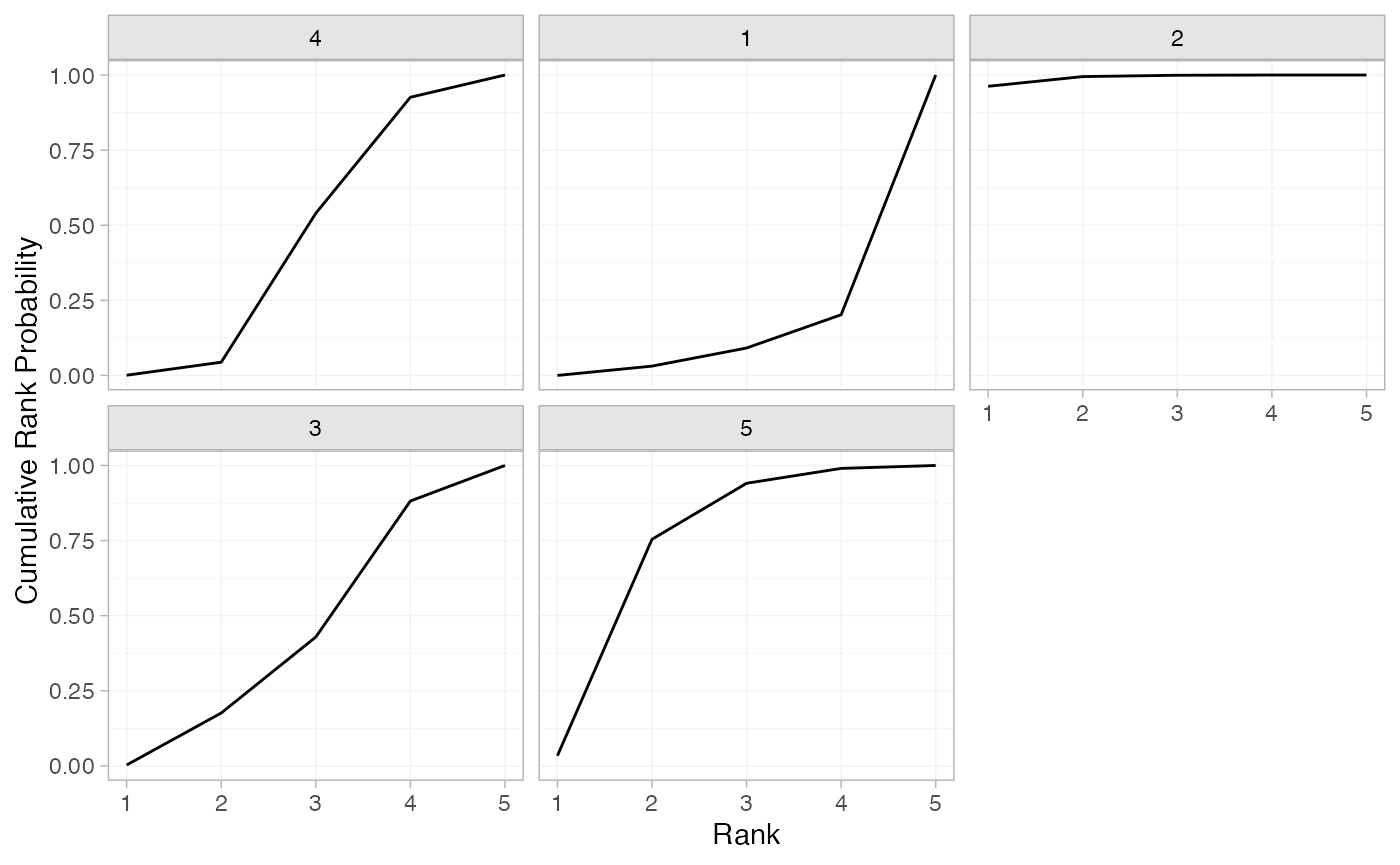
Analysis of mixed arm-based and contrast-based data
We now perform an analysis where some studies contribute arm-based data, and other contribute contrast-based data. Replicating Dias et al. (2011), we consider arm-based data from studies 1-3, and contrast-based data from studies 4-7.
studies <- parkinsons$studyn
(parkinsons_arm <- parkinsons[studies %in% 1:3, ])
#> studyn trtn y se n diff se_diff
#> 1 1 1 -1.22 0.504 54 NA 0.504
#> 2 1 3 -1.53 0.439 95 -0.31 0.668
#> 3 2 1 -0.70 0.282 172 NA 0.282
#> 4 2 2 -2.40 0.258 173 -1.70 0.382
#> 5 3 1 -0.30 0.505 76 NA 0.505
#> 6 3 2 -2.60 0.510 71 -2.30 0.718
#> 7 3 4 -1.20 0.478 81 -0.90 0.695
(parkinsons_contr <- parkinsons[studies %in% 4:7, ])
#> studyn trtn y se n diff se_diff
#> 8 4 3 -0.24 0.265 128 NA 0.265
#> 9 4 4 -0.59 0.354 72 -0.35 0.442
#> 10 5 3 -0.73 0.335 80 NA 0.335
#> 11 5 4 -0.18 0.442 46 0.55 0.555
#> 12 6 4 -2.20 0.197 137 NA 0.197
#> 13 6 5 -2.50 0.190 131 -0.30 0.274
#> 14 7 4 -1.80 0.200 154 NA 0.200
#> 15 7 5 -2.10 0.250 143 -0.30 0.320Setting up the network
We use the functions set_agd_arm() and
set_agd_contrast() to set up the respective data sources
within the network, and then combine together with
combine_network().
mix_arm_net <- set_agd_arm(parkinsons_arm,
study = studyn,
trt = trtn,
y = y,
se = se,
sample_size = n)
mix_contr_net <- set_agd_contrast(parkinsons_contr,
study = studyn,
trt = trtn,
y = diff,
se = se_diff,
sample_size = n)
mix_net <- combine_network(mix_arm_net, mix_contr_net)
mix_net
#> A network with 3 AgD studies (arm-based), and 4 AgD studies (contrast-based).
#>
#> ------------------------------------------------------- AgD studies (arm-based) ----
#> Study Treatment arms
#> 1 2: 1 | 3
#> 2 2: 1 | 2
#> 3 3: 4 | 1 | 2
#>
#> Outcome type: continuous
#> -------------------------------------------------- AgD studies (contrast-based) ----
#> Study Treatment arms
#> 4 2: 4 | 3
#> 5 2: 4 | 3
#> 6 2: 4 | 5
#> 7 2: 4 | 5
#>
#> Outcome type: continuous
#> ------------------------------------------------------------------------------------
#> Total number of treatments: 5
#> Total number of studies: 7
#> Reference treatment is: 4
#> Network is connectedThe sample_size argument is optional, but enables the
nodes to be weighted by sample size in the network plot.
Plot the network structure.
plot(mix_net, weight_edges = TRUE, weight_nodes = TRUE)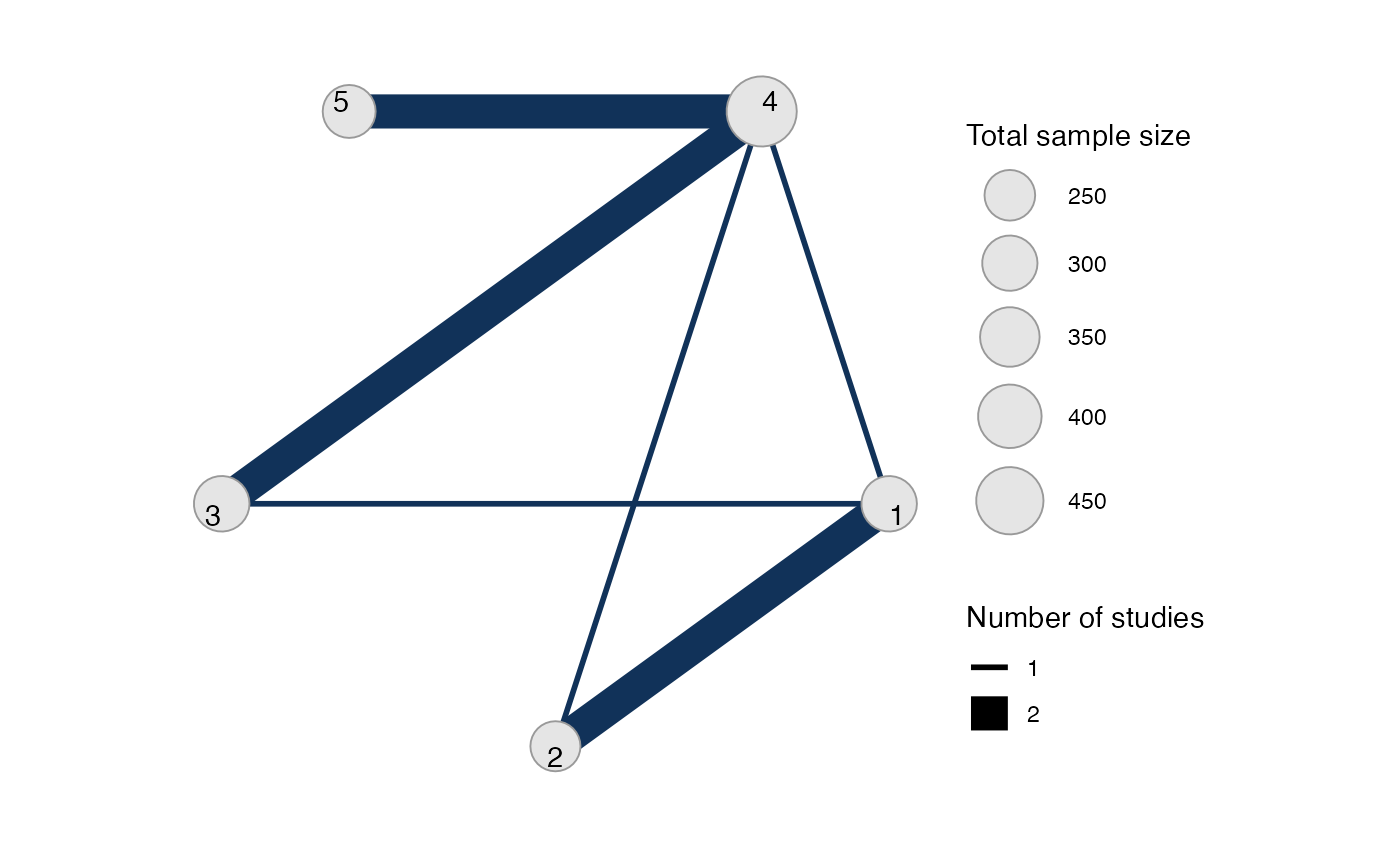
Meta-analysis models
We fit both fixed effect (FE) and random effects (RE) models.
Fixed effect meta-analysis
First, we fit a fixed effect model using the nma()
function with trt_effects = "fixed". We use \mathrm{N}(0, 100^2) prior distributions for
the treatment effects d_k and
study-specific intercepts \mu_j. We can
examine the range of parameter values implied by these prior
distributions with the summary() method:
summary(normal(scale = 100))
#> A Normal prior distribution: location = 0, scale = 100.
#> 50% of the prior density lies between -67.45 and 67.45.
#> 95% of the prior density lies between -196 and 196.The model is fitted using the nma() function.
mix_fit_FE <- nma(mix_net,
trt_effects = "fixed",
prior_intercept = normal(scale = 100),
prior_trt = normal(scale = 100))
#> Note: Setting "4" as the network reference treatment.Basic parameter summaries are given by the print()
method:
mix_fit_FE
#> A fixed effects NMA with a normal likelihood (identity link).
#> Inference for Stan model: normal.
#> 4 chains, each with iter=2000; warmup=1000; thin=1;
#> post-warmup draws per chain=1000, total post-warmup draws=4000.
#>
#> mean se_mean sd 2.5% 25% 50% 75% 97.5% n_eff Rhat
#> d[1] 0.51 0.01 0.48 -0.43 0.18 0.50 0.83 1.46 1586 1
#> d[2] -1.31 0.01 0.53 -2.35 -1.66 -1.31 -0.94 -0.31 1626 1
#> d[3] 0.04 0.01 0.32 -0.62 -0.18 0.04 0.26 0.64 2553 1
#> d[5] -0.30 0.00 0.21 -0.72 -0.44 -0.29 -0.16 0.11 3050 1
#> lp__ -4.66 0.05 1.87 -9.12 -5.73 -4.34 -3.23 -2.01 1640 1
#>
#> Samples were drawn using NUTS(diag_e) at Fri Jun 26 12:38:08 2026.
#> For each parameter, n_eff is a crude measure of effective sample size,
#> and Rhat is the potential scale reduction factor on split chains (at
#> convergence, Rhat=1).By default, summaries of the study-specific intercepts \mu_j are hidden, but could be examined by
changing the pars argument:
The prior and posterior distributions can be compared visually using
the plot_prior_posterior() function:
plot_prior_posterior(mix_fit_FE)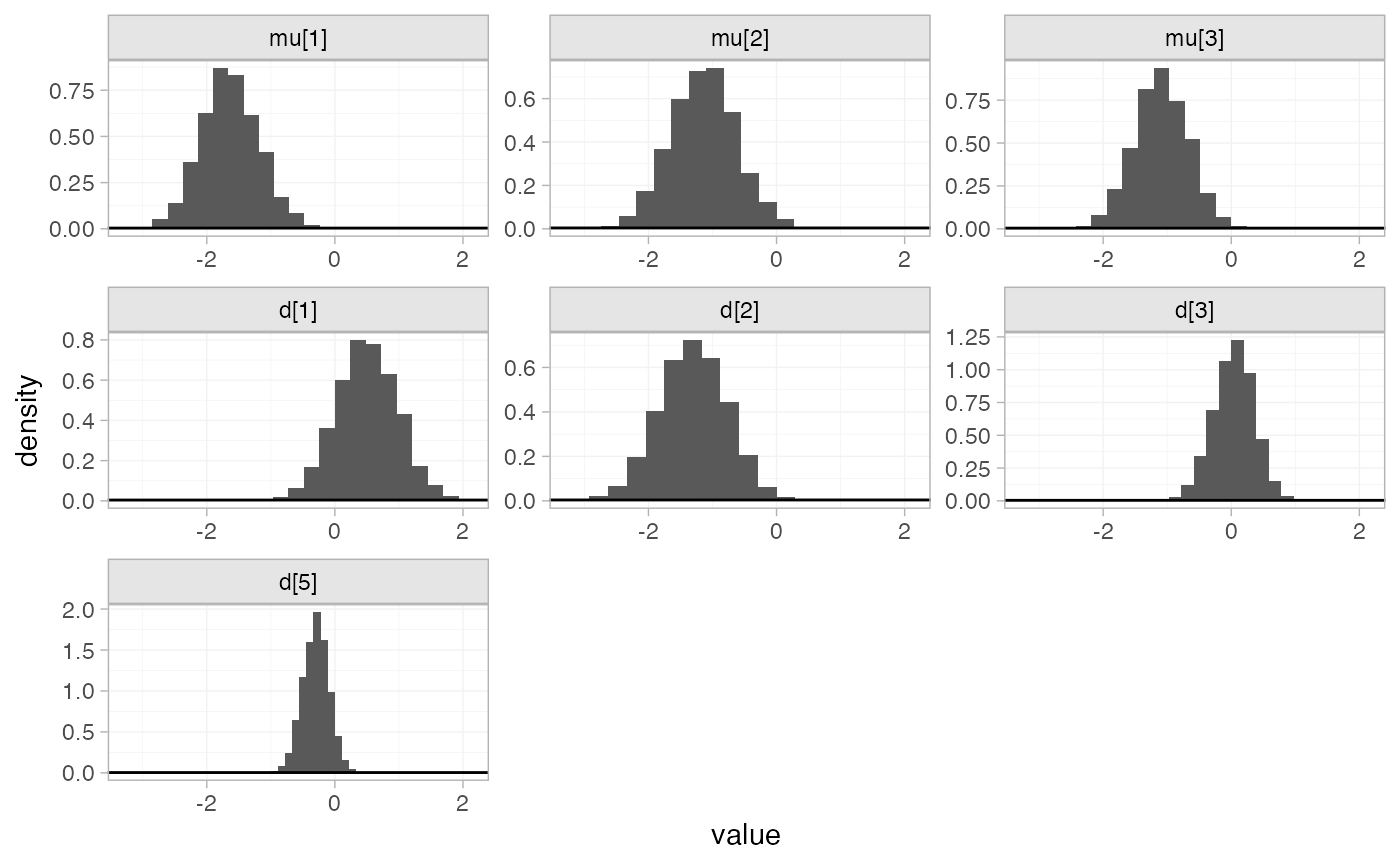
Random effects meta-analysis
We now fit a random effects model using the nma()
function with trt_effects = "random". Again, we use \mathrm{N}(0, 100^2) prior distributions for
the treatment effects d_k and
study-specific intercepts \mu_j, and we
additionally use a \textrm{half-N}(5^2)
prior for the heterogeneity standard deviation \tau. We can examine the range of parameter
values implied by these prior distributions with the
summary() method:
summary(normal(scale = 100))
#> A Normal prior distribution: location = 0, scale = 100.
#> 50% of the prior density lies between -67.45 and 67.45.
#> 95% of the prior density lies between -196 and 196.
summary(half_normal(scale = 5))
#> A half-Normal prior distribution: scale = 5.
#> 50% of the prior density lies between 0 and 3.37.
#> 95% of the prior density lies between 0 and 9.8.Fitting the RE model
mix_fit_RE <- nma(mix_net,
seed = 437219664,
trt_effects = "random",
prior_intercept = normal(scale = 100),
prior_trt = normal(scale = 100),
prior_het = half_normal(scale = 5),
adapt_delta = 0.99)
#> Note: Setting "4" as the network reference treatment.We do see a small number of divergent transition errors, which cannot
simply be removed by increasing the value of the
adapt_delta argument (by default set to 0.95
for RE models). To diagnose, we use the pairs() method to
investigate where in the posterior distribution these divergences are
happening (indicated by red crosses):
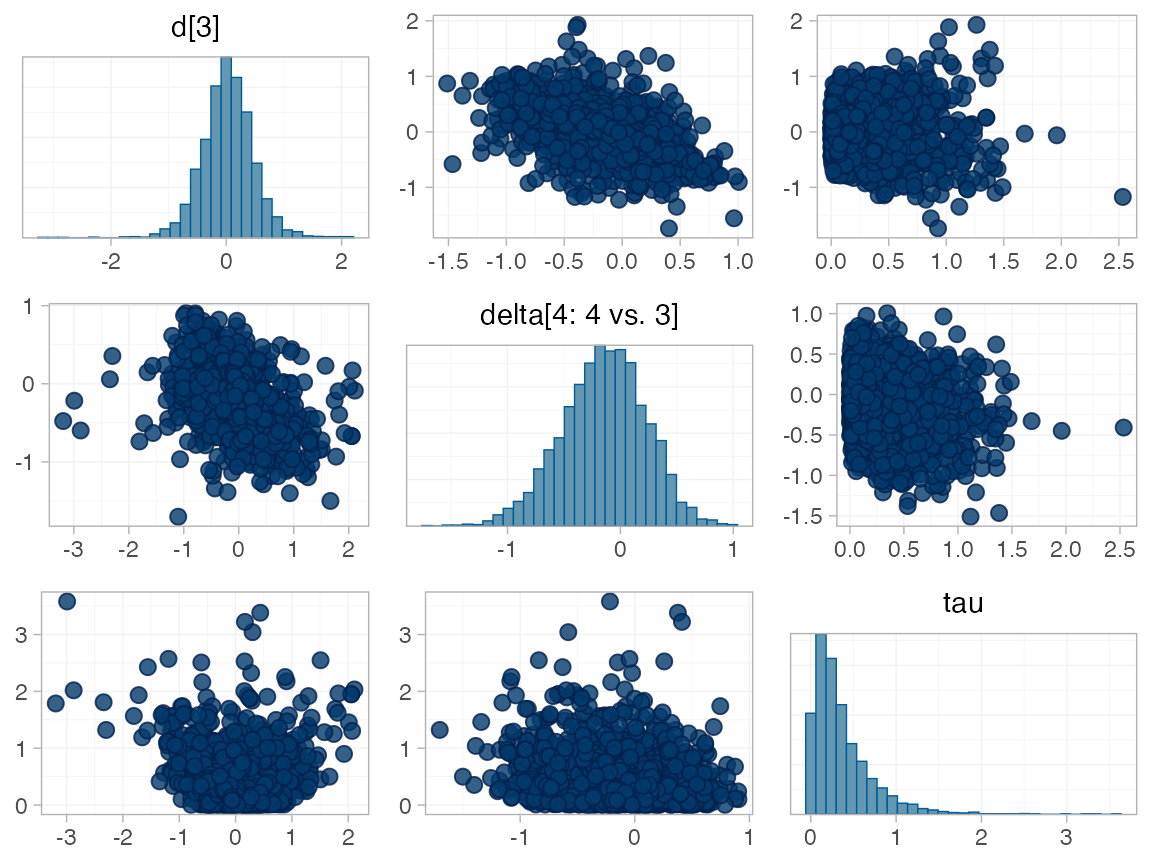
The divergent transitions occur in the upper tail of the heterogeneity standard deviation. In this case, with only a small number of studies, there is not very much information to estimate the heterogeneity standard deviation and the prior distribution may be too heavy-tailed. We could consider a more informative prior distribution for the heterogeneity variance to aid estimation.
Basic parameter summaries are given by the print()
method:
mix_fit_RE
#> A random effects NMA with a normal likelihood (identity link).
#> Inference for Stan model: normal.
#> 4 chains, each with iter=2000; warmup=1000; thin=1;
#> post-warmup draws per chain=1000, total post-warmup draws=4000.
#>
#> mean se_mean sd 2.5% 25% 50% 75% 97.5% n_eff Rhat
#> d[1] 0.55 0.01 0.59 -0.61 0.15 0.54 0.91 1.73 1945 1
#> d[2] -1.30 0.01 0.67 -2.61 -1.73 -1.29 -0.88 0.01 2026 1
#> d[3] 0.03 0.01 0.45 -0.86 -0.24 0.02 0.30 0.92 2923 1
#> d[5] -0.31 0.01 0.40 -1.13 -0.51 -0.30 -0.09 0.47 2330 1
#> lp__ -10.88 0.09 3.26 -18.03 -12.90 -10.60 -8.59 -5.26 1465 1
#> tau 0.37 0.01 0.36 0.01 0.12 0.27 0.49 1.28 1080 1
#>
#> Samples were drawn using NUTS(diag_e) at Fri Jun 26 12:38:10 2026.
#> For each parameter, n_eff is a crude measure of effective sample size,
#> and Rhat is the potential scale reduction factor on split chains (at
#> convergence, Rhat=1).By default, summaries of the study-specific intercepts \mu_j and study-specific relative effects
\delta_{jk} are hidden, but could be
examined by changing the pars argument:
The prior and posterior distributions can be compared visually using
the plot_prior_posterior() function:
plot_prior_posterior(mix_fit_RE)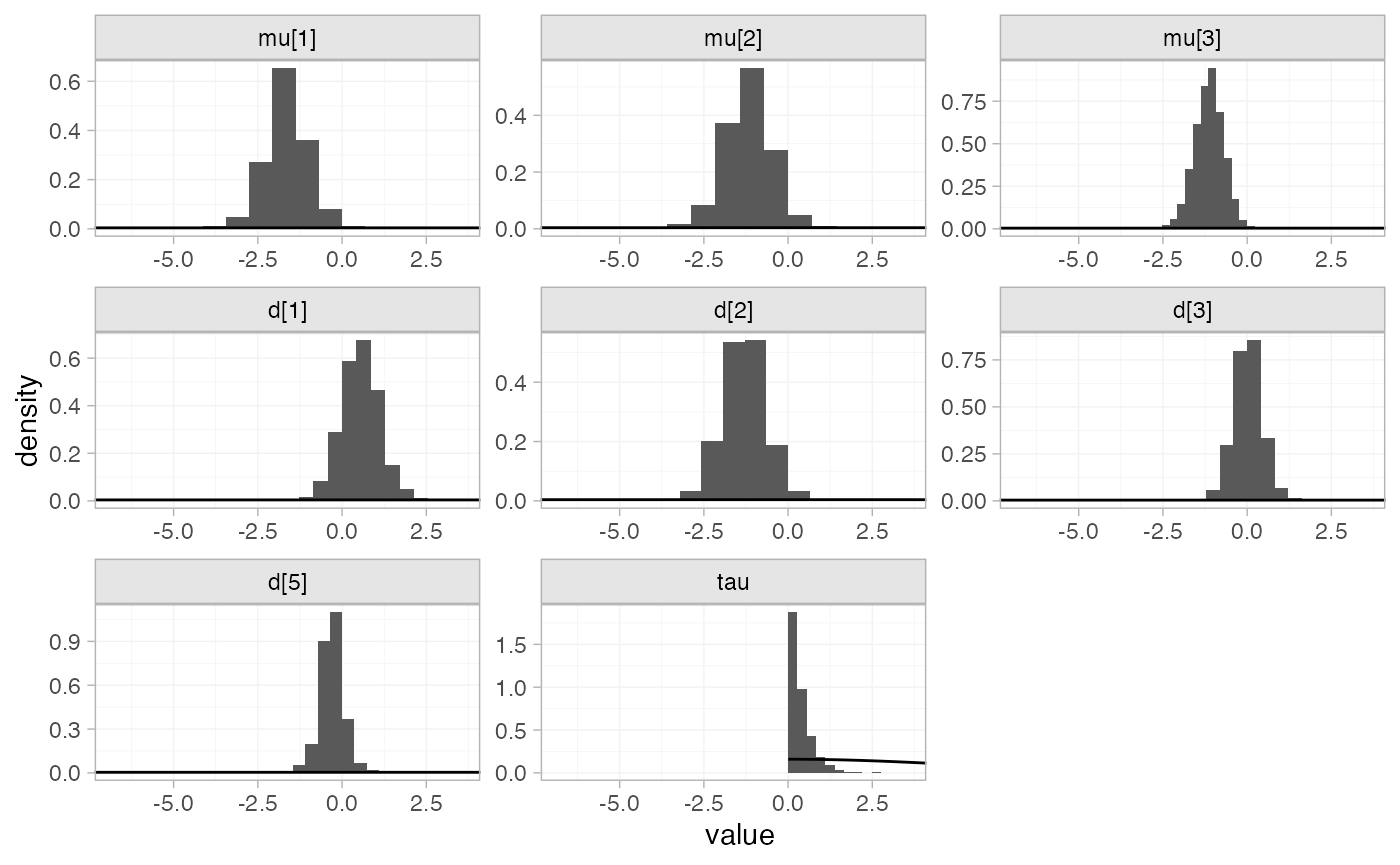
Model comparison
Model fit can be checked using the dic() function:
(mix_dic_FE <- dic(mix_fit_FE))
#> Residual deviance: 9.3 (on 11 data points)
#> pD: 7
#> DIC: 16.3
(mix_dic_RE <- dic(mix_fit_RE))
#> Residual deviance: 9.6 (on 11 data points)
#> pD: 8.4
#> DIC: 18Both models fit the data well, having posterior mean residual deviance close to the number of data points. The DIC is similar between models, so we choose the FE model based on parsimony.
We can also examine the residual deviance contributions with the
corresponding plot() method.
plot(mix_dic_FE)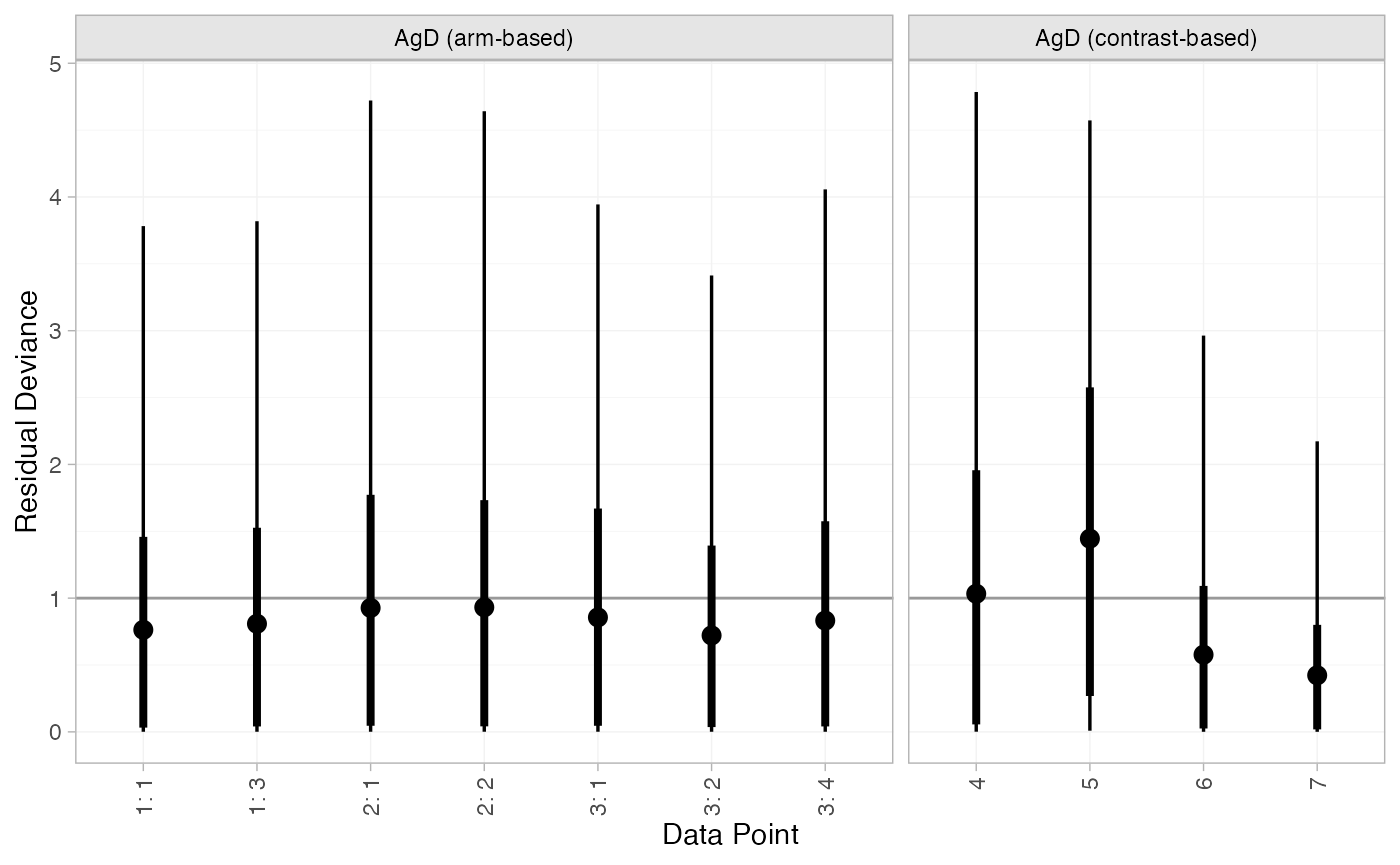
plot(mix_dic_RE)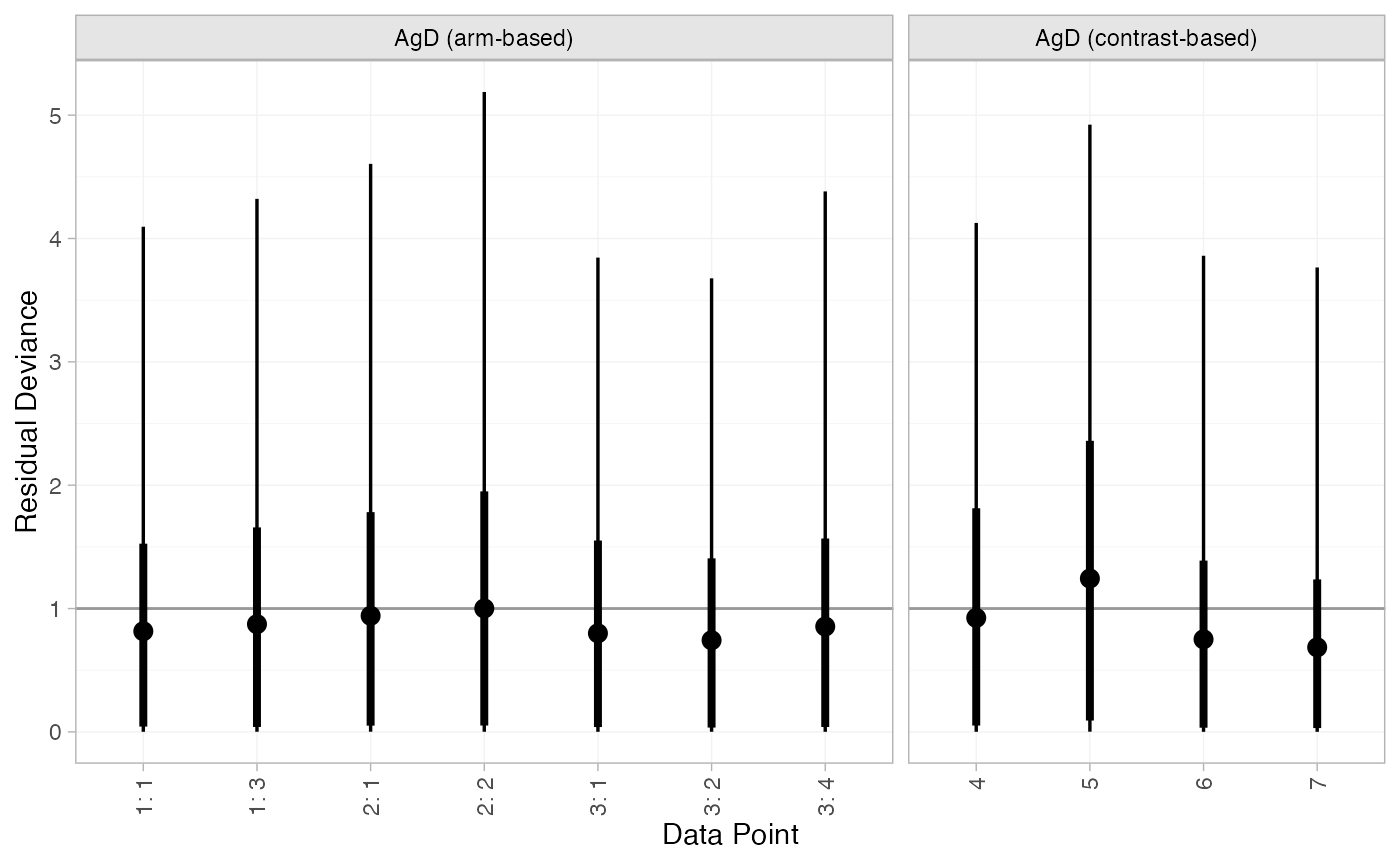
Further results
For comparison with Dias et al. (2011), we can produce relative effects
against placebo using the relative_effects() function with
trt_ref = 1:
(mix_releff_FE <- relative_effects(mix_fit_FE, trt_ref = 1))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> d[4] -0.51 0.48 -1.46 -0.83 -0.50 -0.18 0.43 1606 2262 1
#> d[2] -1.81 0.33 -2.47 -2.04 -1.81 -1.58 -1.16 5739 2842 1
#> d[3] -0.47 0.49 -1.43 -0.80 -0.47 -0.14 0.50 2409 3011 1
#> d[5] -0.80 0.53 -1.85 -1.16 -0.79 -0.45 0.27 1713 2275 1
plot(mix_releff_FE, ref_line = 0)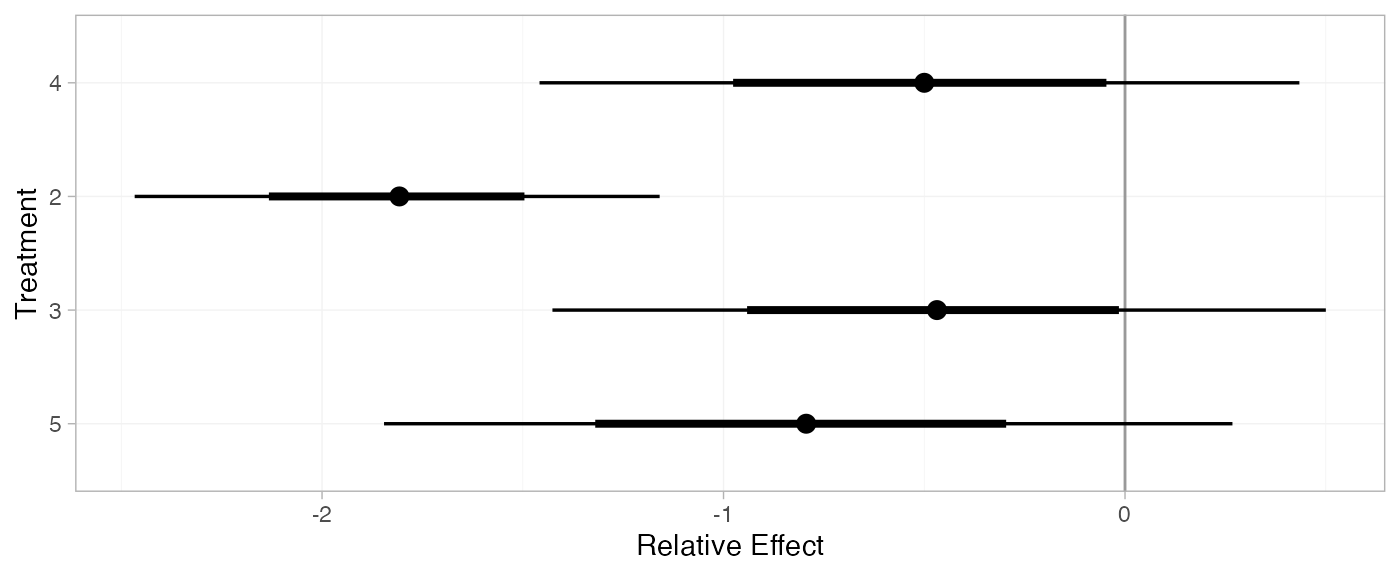
(mix_releff_RE <- relative_effects(mix_fit_RE, trt_ref = 1))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> d[4] -0.55 0.59 -1.73 -0.91 -0.54 -0.15 0.61 1952 2171 1
#> d[2] -1.85 0.49 -2.89 -2.13 -1.85 -1.55 -0.96 4054 2813 1
#> d[3] -0.52 0.62 -1.74 -0.89 -0.51 -0.13 0.64 2989 2480 1
#> d[5] -0.85 0.71 -2.23 -1.28 -0.84 -0.40 0.51 2067 1959 1
plot(mix_releff_RE, ref_line = 0)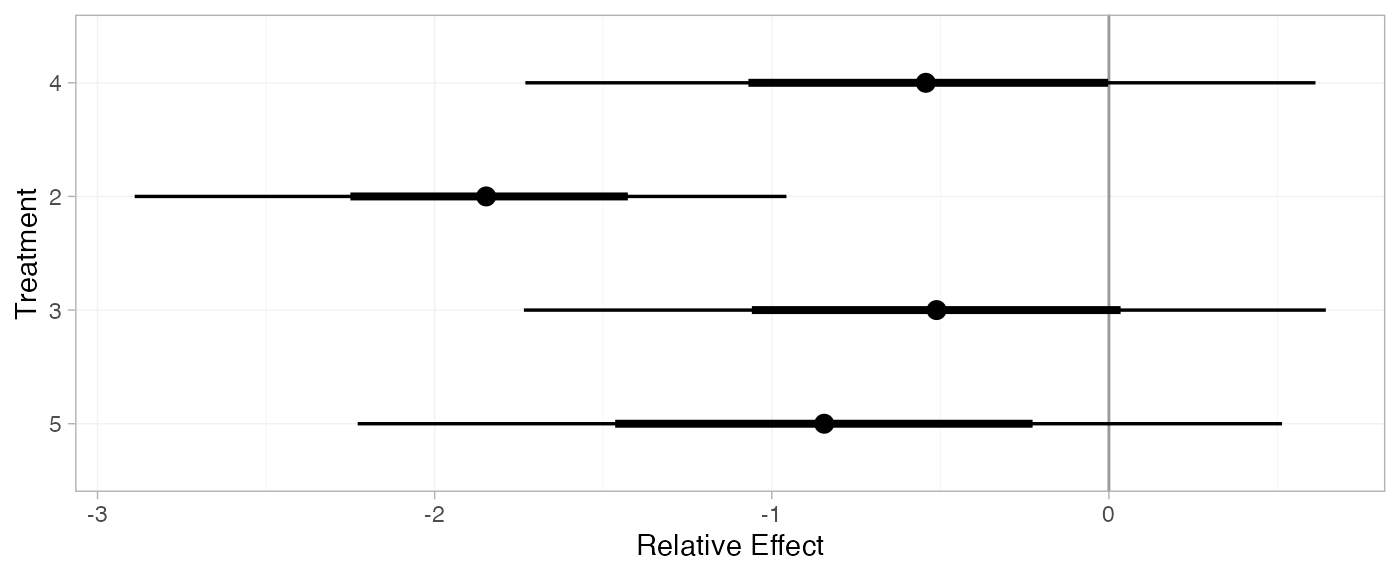
Following Dias et al. (2011), we produce absolute predictions of
the mean off-time reduction on each treatment assuming a Normal
distribution for the outcomes on treatment 1 (placebo) with mean -0.73 and precision 21. We use the predict() method,
where the baseline argument takes a distr()
distribution object with which we specify the corresponding Normal
distribution, and we specify baseline_trt = 1 to indicate
that the baseline distribution corresponds to treatment 1. (Strictly
speaking, type = "response" is unnecessary here, since the
identity link function was used.)
mix_pred_FE <- predict(mix_fit_FE,
baseline = distr(qnorm, mean = -0.73, sd = 21^-0.5),
type = "response",
baseline_trt = 1)
mix_pred_FE
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4] -1.23 0.53 -2.27 -1.59 -1.22 -0.88 -0.17 1777 2613 1
#> pred[1] -0.72 0.22 -1.14 -0.87 -0.72 -0.58 -0.29 3556 3638 1
#> pred[2] -2.53 0.39 -3.31 -2.80 -2.53 -2.26 -1.78 5108 3551 1
#> pred[3] -1.19 0.54 -2.24 -1.55 -1.21 -0.82 -0.10 2525 3106 1
#> pred[5] -1.52 0.57 -2.66 -1.91 -1.52 -1.14 -0.37 1842 2382 1
plot(mix_pred_FE)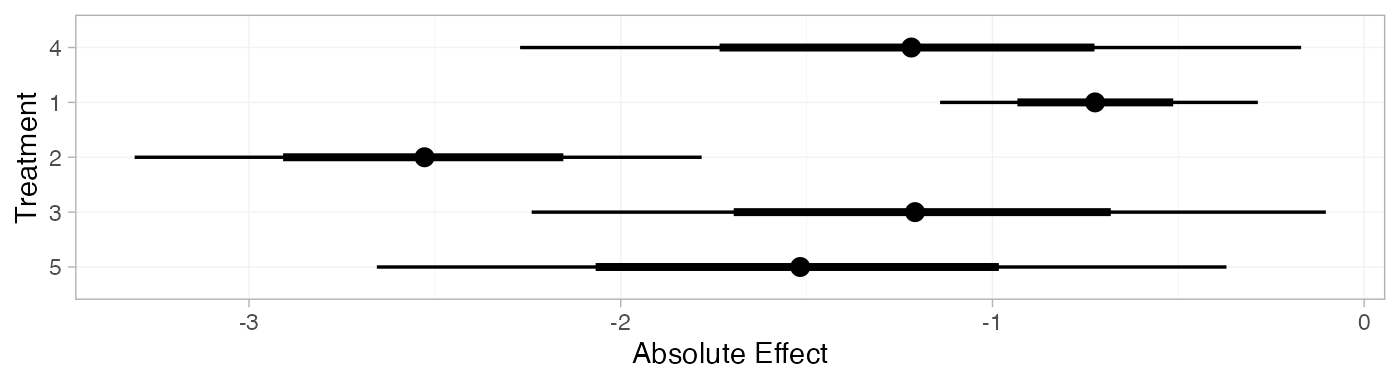
mix_pred_RE <- predict(mix_fit_RE,
baseline = distr(qnorm, mean = -0.73, sd = 21^-0.5),
type = "response",
baseline_trt = 1)
mix_pred_RE
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[4] -1.27 0.63 -2.52 -1.68 -1.27 -0.87 -0.06 2088 2415 1
#> pred[1] -0.73 0.22 -1.17 -0.88 -0.73 -0.58 -0.31 4051 3817 1
#> pred[2] -2.58 0.54 -3.68 -2.90 -2.57 -2.24 -1.57 4000 2896 1
#> pred[3] -1.25 0.65 -2.52 -1.66 -1.24 -0.84 0.01 3099 2869 1
#> pred[5] -1.58 0.74 -2.99 -2.04 -1.58 -1.14 -0.13 2124 2509 1
plot(mix_pred_RE)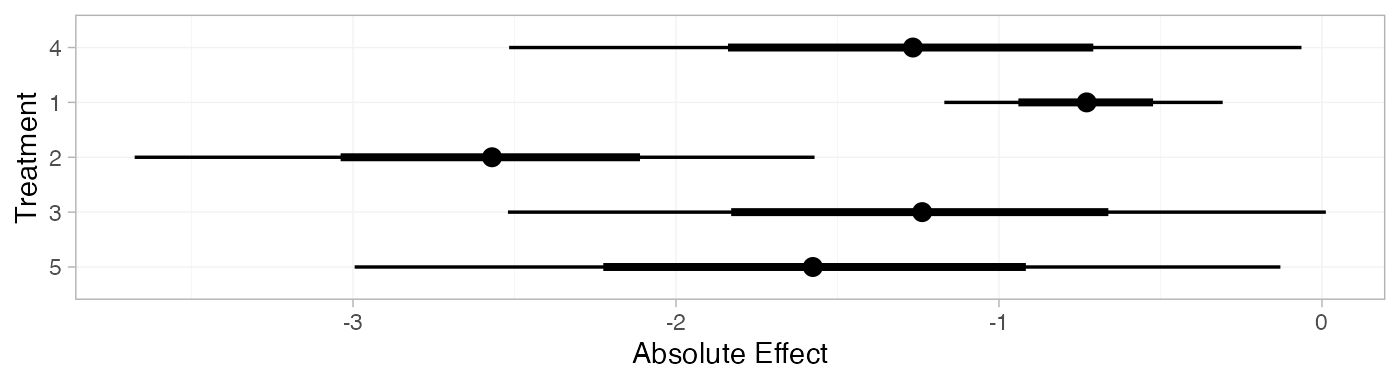
If the baseline argument is omitted, predictions of mean
off-time reduction will be produced for every arm-based study
in the network based on their estimated baseline response \mu_j:
mix_pred_FE_studies <- predict(mix_fit_FE, type = "response")
mix_pred_FE_studies
#> ---------------------------------------------------------------------- Study: 1 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[1: 4] -1.64 0.45 -2.50 -1.94 -1.65 -1.34 -0.70 2068 2403 1
#> pred[1: 1] -1.13 0.43 -1.95 -1.42 -1.14 -0.84 -0.29 3407 2920 1
#> pred[1: 2] -2.94 0.52 -3.93 -3.29 -2.94 -2.59 -1.96 3148 3019 1
#> pred[1: 3] -1.60 0.39 -2.35 -1.86 -1.61 -1.34 -0.84 3552 3294 1
#> pred[1: 5] -1.93 0.50 -2.88 -2.28 -1.93 -1.60 -0.94 2099 2516 1
#>
#> ---------------------------------------------------------------------- Study: 2 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[2: 4] -1.14 0.52 -2.14 -1.50 -1.13 -0.79 -0.10 1589 2167 1
#> pred[2: 1] -0.64 0.26 -1.15 -0.81 -0.63 -0.46 -0.11 4656 3462 1
#> pred[2: 2] -2.45 0.24 -2.93 -2.61 -2.45 -2.28 -1.96 5173 3692 1
#> pred[2: 3] -1.11 0.53 -2.15 -1.46 -1.11 -0.74 -0.05 2303 2812 1
#> pred[2: 5] -1.44 0.56 -2.53 -1.83 -1.44 -1.05 -0.31 1690 2295 1
#>
#> ---------------------------------------------------------------------- Study: 3 ----
#>
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> pred[3: 4] -1.10 0.42 -1.94 -1.38 -1.10 -0.81 -0.28 2007 2612 1
#> pred[3: 1] -0.59 0.36 -1.30 -0.84 -0.60 -0.35 0.11 4337 2979 1
#> pred[3: 2] -2.41 0.39 -3.17 -2.66 -2.41 -2.14 -1.66 3882 3167 1
#> pred[3: 3] -1.06 0.47 -1.99 -1.39 -1.04 -0.75 -0.14 3112 2988 1
#> pred[3: 5] -1.40 0.48 -2.34 -1.71 -1.40 -1.06 -0.47 2072 2409 1
plot(mix_pred_FE_studies)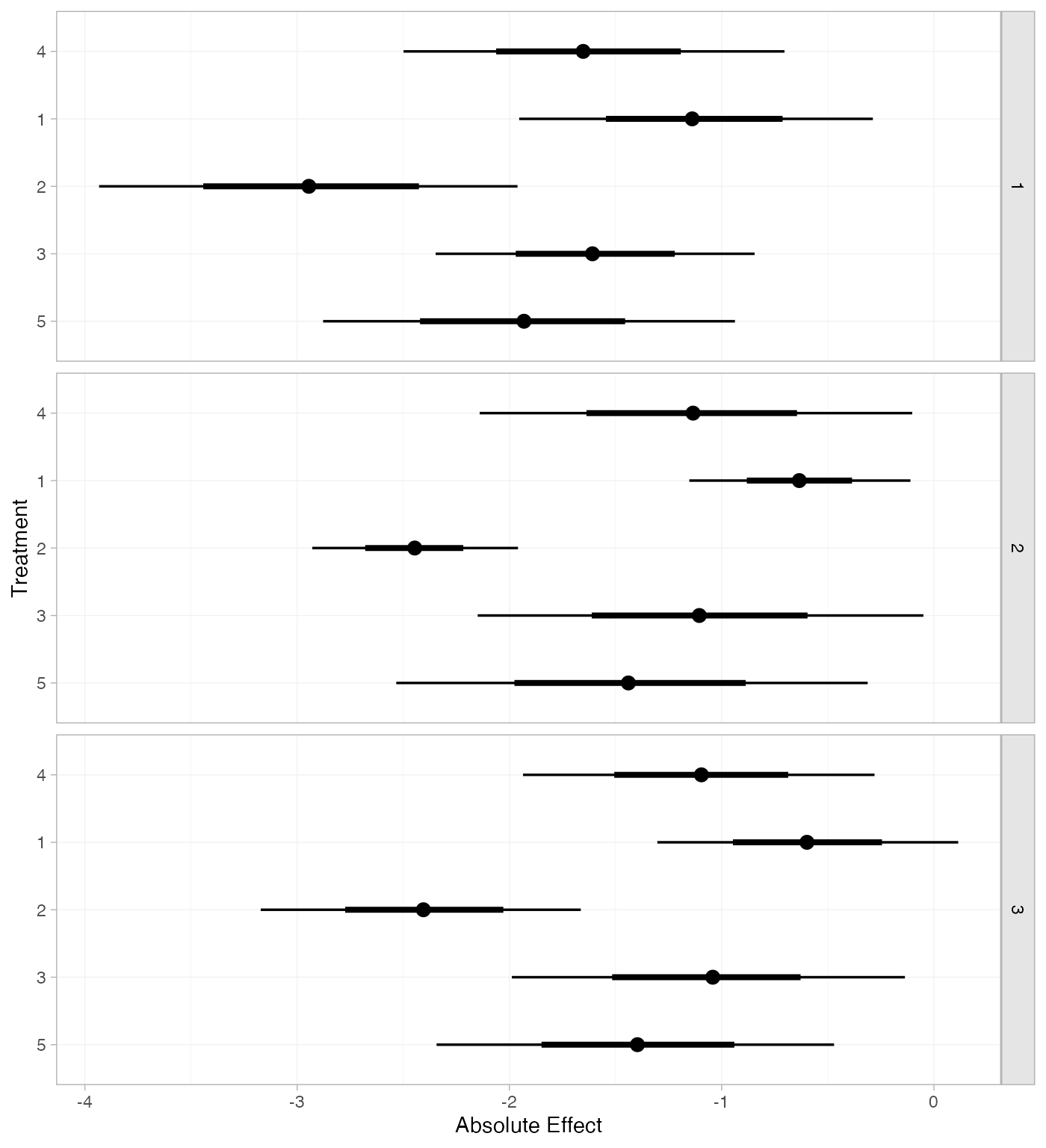
We can also produce treatment rankings, rank probabilities, and cumulative rank probabilities.
(mix_ranks <- posterior_ranks(mix_fit_FE))
#> mean sd 2.5% 25% 50% 75% 97.5% Bulk_ESS Tail_ESS Rhat
#> rank[4] 3.51 0.71 2 3 3 4 5 2379 NA 1
#> rank[1] 4.63 0.79 2 5 5 5 5 2077 NA 1
#> rank[2] 1.05 0.26 1 1 1 1 2 2572 2682 1
#> rank[3] 3.52 0.92 2 3 4 4 5 3397 NA 1
#> rank[5] 2.29 0.68 1 2 2 3 4 2517 2745 1
plot(mix_ranks)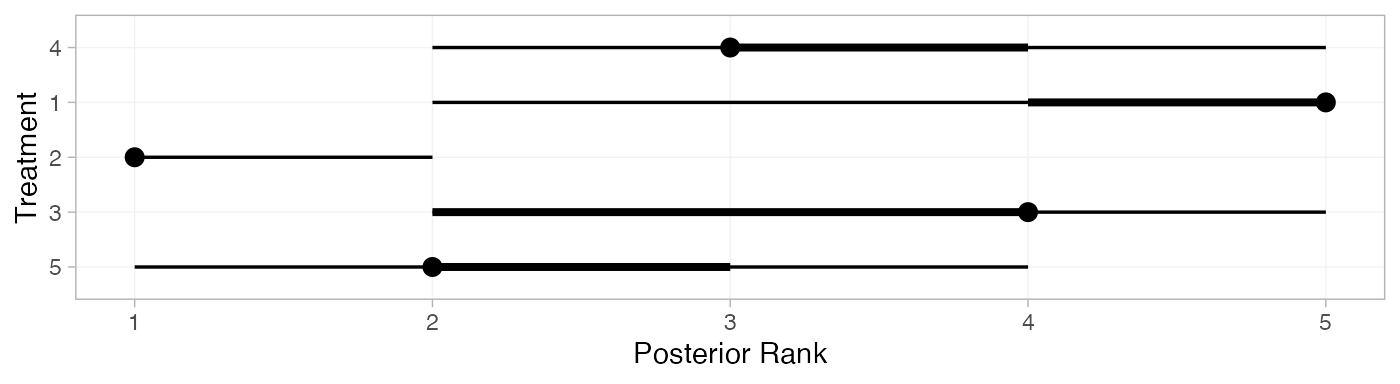
(mix_rankprobs <- posterior_rank_probs(mix_fit_FE))
#> p_rank[1] p_rank[2] p_rank[3] p_rank[4] p_rank[5]
#> d[4] 0.00 0.04 0.49 0.39 0.08
#> d[1] 0.00 0.04 0.07 0.11 0.78
#> d[2] 0.96 0.03 0.01 0.00 0.00
#> d[3] 0.00 0.17 0.25 0.45 0.13
#> d[5] 0.04 0.71 0.19 0.05 0.01
plot(mix_rankprobs)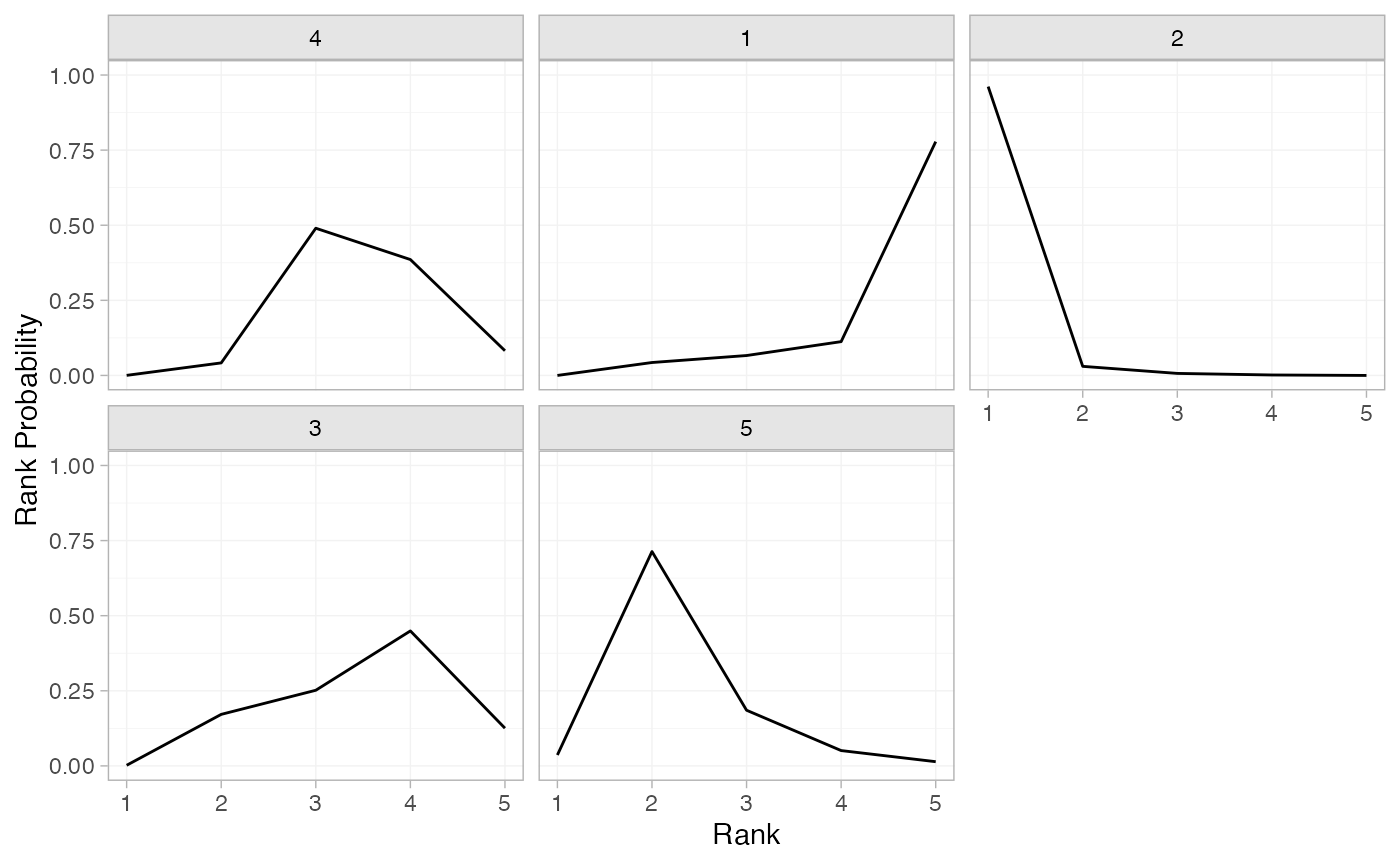
(mix_cumrankprobs <- posterior_rank_probs(mix_fit_FE, cumulative = TRUE))
#> p_rank[1] p_rank[2] p_rank[3] p_rank[4] p_rank[5]
#> d[4] 0.00 0.04 0.53 0.92 1
#> d[1] 0.00 0.04 0.11 0.22 1
#> d[2] 0.96 0.99 1.00 1.00 1
#> d[3] 0.00 0.17 0.43 0.87 1
#> d[5] 0.04 0.75 0.94 0.99 1
plot(mix_cumrankprobs)